


Avances en el diagnóstico y tratamiento de la fibrosis quística: Un análisis exhaustivo en la era de la medicina de precisión
José Hernández Jiménez
9/8/202517 min leer
El Paisaje de la Fibrosis Quística: Del Gen a la Disfunción Molecular
La fibrosis quística (FQ) se ha redefinido en la última década, pasando de ser una enfermedad mortal en la infancia a una condición crónica manejable para muchos pacientes. Esta transformación ha sido impulsada por una comprensión cada vez más profunda de su base molecular y una consecuente revolución en el desarrollo de terapias dirigidas. El núcleo de esta enfermedad reside en la disfunción de una sola proteína, el regulador de conductancia transmembrana de la fibrosis quística (CFTR).
Una Fisiopatología Redefinida: La Centralidad del Canal CFTR
La fibrosis quística es una enfermedad monogénica que afecta a las células epiteliales de múltiples órganos, principalmente los pulmones, el páncreas, el hígado y los intestinos. La patología sistémica de la FQ surge de la incapacidad de las células epiteliales para transportar iones de cloruro y bicarbonato a través de sus membranas apicales. Esta función es normalmente llevada a cabo por la proteína CFTR, que actúa como un canal iónico regulado. Cuando el canal CFTR es disfuncional, el transporte de iones está severamente comprometido, lo que resulta en una deshidratación de las secreciones mucosas. Este moco espeso y pegajoso obstruye los conductos de los órganos, dando lugar a la manifestación clínica de la enfermedad, que incluye enfermedad pulmonar obstructiva crónica, insuficiencia pancreática y enfermedad hepática.
Los avances en la comprensión de la función de la proteína CFTR no han sido meramente una curiosidad académica. Esta comprensión detallada de la fisiopatología ha sentado las bases para un cambio de paradigma terapéutico. En lugar de tratar únicamente los síntomas de la enfermedad, como las infecciones pulmonares recurrentes o la malabsorción, el campo se ha movido hacia un enfoque de medicina de precisión. Este nuevo enfoque busca corregir la disfunción proteica subyacente, lo que representa un cambio fundamental en la estrategia de tratamiento. Este cambio de una visión macroscópica y sintomática a una visión molecular y genética ha sido la fuerza impulsora detrás de los avances más significativos en la atención de la FQ.
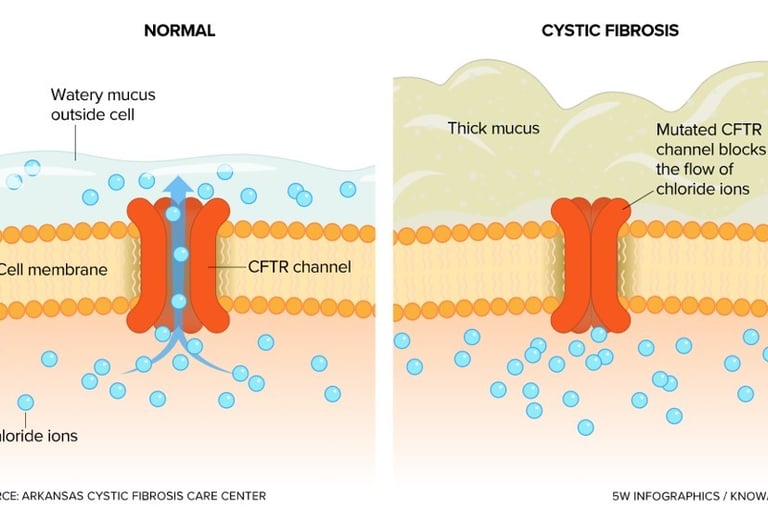
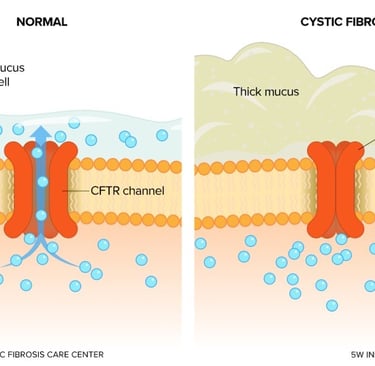
imagen de: ivd.palexmedical.com
El Espectro Genético del CFTR: Mutaciones y su Consecuencia Funcional
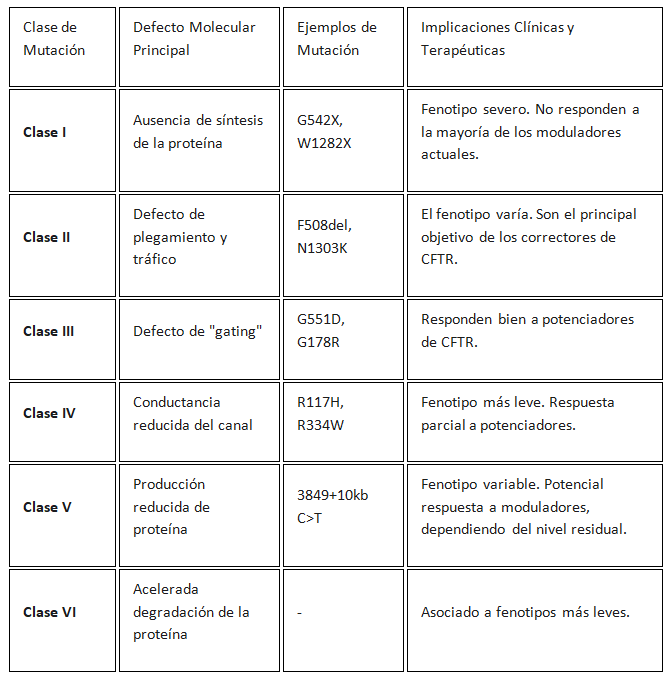
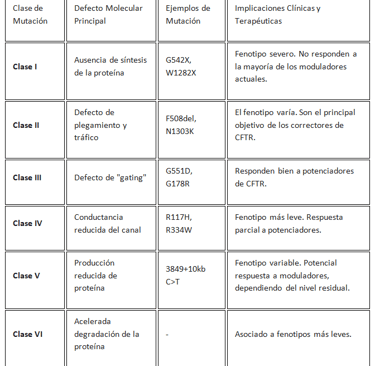
El factor etiológico de la fibrosis quística es una mutación en el gen CFTR, localizado en el cromosoma 7. Más de 2,000 mutaciones se han identificado en este gen, pero las mutaciones clínicamente relevantes se clasifican en un sistema de clases que se correlaciona directamente con la gravedad de la disfunción proteica. Esta clasificación es fundamental para el desarrollo y la aplicación de terapias dirigidas. Las clases son:
· Clase I: Mutaciones de terminación que resultan en la ausencia completa de síntesis de la proteína CFTR.
· Clase II: Mutaciones que causan un defecto en el plegamiento y el tráfico de la proteína. La proteína se produce, pero es incorrectamente procesada por el retículo endoplasmático y degradada antes de llegar a la membrana celular. La mutación F508del es el ejemplo más común de esta clase y es la mutación más frecuente en todo el mundo.
· Clase III: Mutaciones de "gating" que permiten que la proteína alcance la membrana celular, pero con un defecto en la apertura del canal. El canal no se activa correctamente, impidiendo el flujo de iones.
· Clase IV: Mutaciones que resultan en una conductancia reducida del canal.
· Clase V: Mutaciones que producen una cantidad reducida de proteína CFTR funcional.
· Clase VI: Mutaciones que causan una acelerada degradación de la proteína en la membrana.
El entendimiento detallado de estas clases de mutaciones ha sido la base para el diseño de moduladores que se dirigen a defectos específicos. Esto demuestra cómo la clasificación funcional de las mutaciones es un requisito indispensable para la revolución de la medicina de precisión.
Más Allá del CFTR: El Rol de los Genes Modificadores
La correlación entre el genotipo y el fenotipo en la FQ no es completamente lineal; la misma mutación puede manifestarse con una gravedad variable en diferentes individuos. Esta variabilidad fenotípica se explica, en parte, por el papel de los "genes modificadores." Estos genes no causan la enfermedad por sí mismos, pero influyen en la expresión y gravedad de la patología causada por la mutación del CFTR. El estudio de estos genes modificadores es un área de investigación activa, ya que podrían explicar por qué algunos pacientes con mutaciones típicamente graves tienen un curso de enfermedad más leve, o viceversa. Esto abre una nueva vía de investigación para la medicina de precisión, más allá de la simple corrección de la proteína CFTR.
La Evolución del Diagnóstico
El diagnóstico de la FQ ha evolucionado significativamente, pasando de una confirmación reactiva de la enfermedad a una detección proactiva y funcional. Esta transformación refleja la creciente importancia de la detección temprana para permitir intervenciones que alteren el curso natural de la enfermedad.
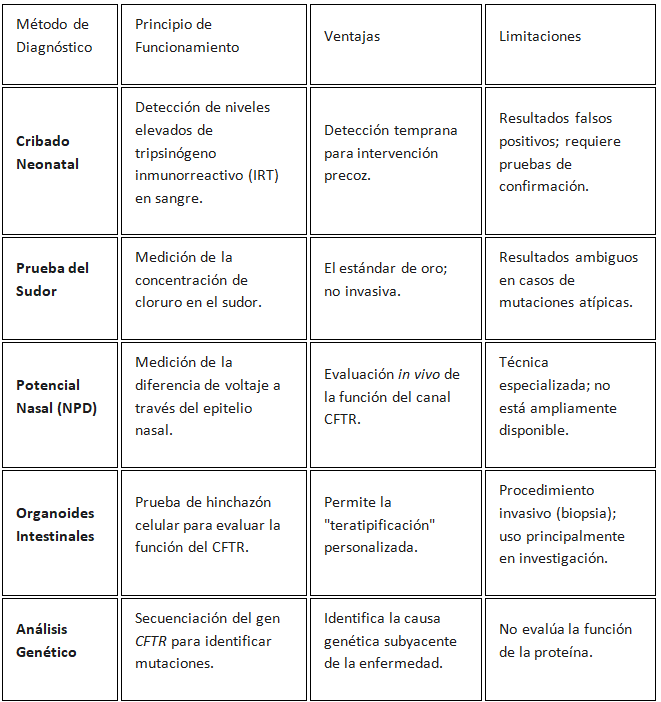
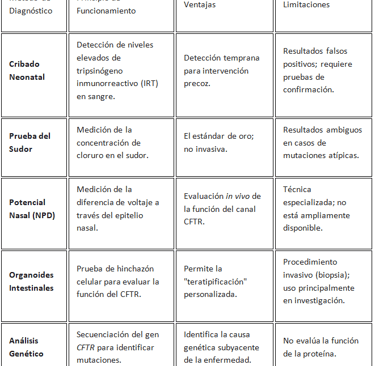
Cribado Neonatal: La Vanguardia de la Detección Temprana
El cribado neonatal (NBS) para la FQ es un pilar de la salud pública moderna, permitiendo la detección de la enfermedad en los primeros días de vida. Este enfoque ha mejorado drásticamente los resultados a largo plazo de los pacientes, ya que el diagnóstico temprano facilita la implementación de un manejo clínico intensivo antes de que ocurra un daño irreversible. El cribado neonatal global varía en su implementación y metodologías, con diferentes países adoptando distintas estrategias de detección. A un resultado positivo en el cribado neonatal le sigue una cascada diagnóstica que debe ser confirmada con pruebas adicionales, como la prueba del sudor, para establecer un diagnóstico definitivo.
Biomarcadores Fisiológicos: Pruebas del Sudor y Potencial Nasal
La prueba del sudor, que mide la concentración de cloruro en la piel, sigue siendo el "estándar de oro" para el diagnóstico de la FQ. Una concentración elevada de cloruro en el sudor es una indicación clave de la disfunción del canal CFTR. Sin embargo, la prueba tiene limitaciones, ya que ciertos pacientes con mutaciones atípicas pueden tener resultados intermedios. Para estos casos, la medición del potencial nasal (NPD) se ha convertido en un biomarcador fisiológico complementario. El NPD mide la diferencia de voltaje a través del epitelio nasal, proporcionando una evaluación in vivo de la actividad del canal CFTR. Su capacidad para detectar la disfunción del CFTR la hace particularmente valiosa en pacientes con resultados de la prueba del sudor ambiguos.
Nuevas Fronteras: Diagnóstico Molecular y Funcional
El diagnóstico de la FQ en el siglo XXI no se limita a las pruebas fisiológicas. La era de la medicina de precisión exige métodos que no solo confirmen la enfermedad, sino que también caractericen la disfunción proteica con una precisión sin precedentes.
· Organoides Intestinales: La tecnología de organoides ha emergido como una herramienta innovadora. Los organoides son estructuras celulares 3D derivadas de biopsias rectales del paciente. Estas estructuras pueden usarse para una técnica conocida como "teratipificación", donde se evalúa la respuesta del tejido del paciente a diferentes moduladores del CFTR. La funcionalidad del canal CFTR se mide a través del grado de hinchazón de los organoides en respuesta a la forskolina. Una respuesta de hinchazón positiva en organoides tratados con moduladores se correlaciona con una respuesta clínica favorable en los pacientes. El uso de organoides ha cambiado el diagnóstico de un proceso de confirmación de la enfermedad a un paso integrado en la estrategia terapéutica.
· Biomarcadores Moleculares y la Inteligencia Artificial: La investigación se está moviendo hacia el uso de biomarcadores para el diagnóstico y el monitoreo de la enfermedad. La metabolómica, por ejemplo, está siendo explorada para identificar biomarcadores predictivos de la FQ. Además, el análisis del aliento exhalado está demostrando ser una técnica prometedora para la detección de patógenos pulmonares como Pseudomonas aeruginosa. La inteligencia artificial (IA) también está siendo evaluada como una herramienta para la detección temprana y el análisis de datos complejos, especialmente en el contexto de enfermedades raras como la FQ.
La evolución del diagnóstico de la FQ demuestra un cambio fundamental de simplemente detectar la presencia de una enfermedad a diagnosticar el defecto funcional específico en cada paciente. Este enfoque es crucial para el despliegue exitoso de las terapias de precisión.
Correlaciones Genotipo-Fenotipo y Heterogeneidad Clínica
A pesar de ser una enfermedad monogénica, la FQ presenta una notable variabilidad en su fenotipo clínico. La comprensión de esta heterogeneidad es esencial para el manejo clínico, especialmente en la era de los moduladores del CFTR.
Variabilidad del Fenotipo: Más Allá de las Mutaciones Clásicas
Dos pacientes con las mismas mutaciones en el gen CFTR pueden experimentar diferentes niveles de gravedad de la enfermedad y afectación de órganos. Esta variabilidad se debe a una combinación de factores genéticos adicionales y ambientales. El concepto de "genes modificadores" explica por qué la misma mutación puede llevar a un fenotipo pulmonar más o menos grave, o a una afectación pancreática variable. Por ejemplo, se han identificado genes modificadores que pueden influir en el fenotipo de la enfermedad hepática, lo que sugiere que la enfermedad es el resultado de la interacción entre la mutación primaria en CFTR y un complejo de otros factores genéticos.
Genética Poblacional: Diferencias Étnicas en el Espectro de Mutaciones
La frecuencia y el espectro de las mutaciones del gen CFTR varían considerablemente entre diferentes poblaciones y grupos étnicos. Mientras que la mutación F508del es extremadamente prevalente en las poblaciones caucásicas, su frecuencia es mucho menor en otras etnias, como en las poblaciones rusas y bielorrusas. Estudios de poblaciones específicas, como la de Bashkortostán, han revelado perfiles de mutaciones únicos, lo que subraya la necesidad de enfoques de cribado y diagnóstico personalizados basados en la genética de la población local. La identificación de estas variaciones es crucial para optimizar los programas de cribado neonatal y para desarrollar terapias dirigidas que se adapten a las mutaciones prevalentes en cada región. Esta heterogeneidad genética de la FQ a nivel global tiene un impacto directo en las estrategias de salud pública y en la industria farmacéutica.
Nuevos Desafíos Clínicos: Manejo de Pacientes que Envejecen
El éxito de las nuevas terapias ha alterado fundamentalmente la historia natural de la FQ. La esperanza de vida de los pacientes ha aumentado drásticamente, lo que ha transformado la FQ de una enfermedad pediátrica a una condición que requiere un manejo a lo largo de toda la vida adulta. Este cambio ha creado un nuevo conjunto de desafíos clínicos que no existían en el pasado.
A medida que los pacientes con FQ envejecen, comienzan a desarrollar comorbilidades que son comunes en la población general, como la diabetes, la osteoporosis y la enfermedad renal crónica. Esto obliga a los médicos a reevaluar y adaptar las guías de manejo clínico para incorporar el cuidado de enfermedades crónicas que van más allá del enfoque tradicional en el sistema pulmonar y digestivo. Además, la salud reproductiva ha emergido como un área de creciente importancia. Los estudios han abordado las consideraciones para el embarazo en mujeres con FQ y han evaluado la salud sexual y la fertilidad en pacientes masculinos. La prolongación de la esperanza de vida de los pacientes es un testimonio del éxito de las terapias, pero a su vez, exige una evolución completa del modelo de atención médica para abordar los desafíos de una población que envejece con una enfermedad genética compleja.
El Cambio de Paradigma en el Tratamiento: La Revolución de los Moduladores del CFTR
La introducción de los moduladores del CFTR representa el avance terapéutico más significativo en la historia de la fibrosis quística. Estas terapias no se limitan a tratar los síntomas, sino que abordan la causa molecular subyacente de la enfermedad, marcando una verdadera transición a la medicina de precisión.
Mecanismos de Acción de la Terapia Moduladora
Los moduladores del CFTR son pequeñas moléculas diseñadas para corregir los defectos específicos de la proteína CFTR, dependiendo de la clase de mutación. Se clasifican en diferentes categorías según su función:
· Potenciadores (Potentiators): Estas moléculas, como ivacaftor, actúan sobre la proteína CFTR en la membrana celular. Su función es aumentar la probabilidad de apertura del canal iónico, permitiendo un mayor flujo de iones de cloruro a través de la membrana. Son particularmente eficaces en pacientes con mutaciones de "gating" (Clase III).
· Correctores (Correctors): Estas moléculas, como elexacaftor y tezacaftor, ayudan a la proteína CFTR mutada (típicamente F508del) a plegarse correctamente en el retículo endoplasmático y a ser transportada de manera eficiente a la membrana celular. Abordan el defecto de tráfico (Clase II), la mutación más común.
· Amplificadores (Amplifiers): Esta es una clase más nueva de moduladores que tiene como objetivo aumentar la cantidad total de proteína CFTR producida.
La existencia de estos diferentes tipos de moduladores es una demostración directa de cómo una comprensión detallada de las clases de mutaciones se tradujo directamente en el desarrollo de terapias dirigidas.
El Impacto de las Terapias Combinadas Altamente Efectivas (HEMT)
El desarrollo de terapias combinadas, conocidas como terapias de modulación altamente efectivas (HEMT, por sus siglas en inglés), ha sido un hito. La terapia de triple combinación, elexacaftor/tezacaftor/ivacaftor, ha demostrado un impacto clínico significativo. Estudios han mostrado mejoras notables en la función pulmonar, evaluada por el volumen espiratorio forzado en un segundo (FEV1), y una reducción en la tasa de exacerbaciones pulmonares. Este impacto no solo se ha reflejado en las métricas clínicas, sino también en una mejoría sustancial en la calidad de vida de los pacientes. La efectividad de estas terapias ha desplazado el enfoque del manejo de la enfermedad de tratar los síntomas a mantener la salud y prevenir complicaciones a largo plazo.
Manejo Clínico en la Era de los Moduladores
La introducción de los moduladores ha cambiado la práctica clínica. Los pacientes que responden a los moduladores pueden experimentar una reducción en la necesidad de los tratamientos tradicionales, como los antibióticos y las terapias de aclaramiento de las vías aéreas. El efecto de los moduladores en la patogénesis subyacente de la enfermedad puede incluso alterar el microbioma pulmonar y el patrón de infecciones bacterianas. Sin embargo, la revolución de los moduladores también ha creado una nueva dicotomía: los pacientes elegibles que responden a las terapias y un grupo significativo de pacientes con mutaciones raras o de Clase I que no se benefician de estas terapias.
La Próxima Generación de Terapias y el Futuro de la FQ
Si bien los moduladores del CFTR han transformado el pronóstico de la enfermedad, la visión a largo plazo para la fibrosis quística se extiende más allá de la terapia con moléculas pequeñas. El siguiente objetivo es una corrección fundamental del defecto genético, lo que transformaría la FQ de una condición crónica a una enfermedad potencialmente curable.
El Amanecer de la Terapia Génica y la Terapia de ARNm
Para los pacientes que no son elegibles para los moduladores, las terapias genéticas y de ARN ofrecen la promesa de una solución definitiva. El campo de la terapia génica busca introducir una copia funcional del gen CFTR en las células de las vías respiratorias para restaurar la producción de proteína normal. Los enfoques de terapia génica incluyen el uso de vectores virales y no virales, cada uno con sus propios desafíos de entrega in vivo y de seguridad.
Una alternativa prometedora son las terapias basadas en ARN. Por ejemplo, la terapia de ARN mensajero (ARNm) tiene como objetivo introducir una plantilla de ARNm funcional en la célula para que la célula pueda producir la proteína CFTR correcta de novo. Los oligonucleótidos antisentido también están siendo explorados para modular la expresión de genes o el empalme (splicing) del ARNm. Estas terapias son lógicamente el siguiente paso para abordar las mutaciones de Clase I que no producen ninguna proteína CFTR. La visión es que, al proporcionar la "instrucción" genética faltante o defectuosa, se puede lograr la producción de una proteína funcional, lo que representa una corrección más radical que la simple reparación de una proteína defectuosa.
Hacia la Medicina Personalizada Completa
El éxito de la terapia con moduladores ha validado el enfoque de la medicina de precisión en la fibrosis quística. El futuro de la investigación se centrará en extender este enfoque a todos los pacientes, incluyendo aquellos con mutaciones raras. El futuro del desarrollo de fármacos para la FQ no se limita a encontrar "una píldora mejor"; se enfoca en abordar la totalidad del panorama genético, incluyendo los genes modificadores que pueden influir en la respuesta al tratamiento. El desarrollo de terapias dirigidas a estas vías genéticas adicionales podría optimizar los resultados para los pacientes y mitigar los efectos secundarios.
Desafíos Persistentes y Recomendaciones para la Investigación Futura
A pesar de los avances transformadores, persisten desafíos significativos que deben abordarse para mejorar la vida de todos los pacientes con FQ.
Superar las Barreras de la Terapia Moduladora
El mayor desafío no resuelto es la población de pacientes con mutaciones raras o con mutaciones de Clase I para quienes no existe una terapia moduladora disponible. Se requiere una inversión continua en la investigación de terapias alternativas, como la terapia génica, para esta población no elegible. Además, a medida que los pacientes permanecen en terapia con moduladores por períodos más largos, es crucial monitorear los efectos secundarios a largo plazo y la adherencia al tratamiento.
El Futuro del Monitoreo y la Investigación Clínica
El éxito de los moduladores ha hecho que las medidas tradicionales de la función pulmonar, como el FEV1, sean menos sensibles para el monitoreo de la progresión de la enfermedad. Hay una necesidad crítica de nuevos biomarcadores no invasivos y sensibles que puedan detectar cambios sutiles en la salud pulmonar. La investigación está explorando biomarcadores de inflamación en el esputo y el aliento exhalado para este propósito. Asimismo, se requieren nuevas guías para los ensayos clínicos y el manejo de la enfermedad, que se adapten a la nueva realidad de una población de pacientes que envejece con una enfermedad controlada.
Referencias:
· Advances in the Cystic Fibrosis Drug Development Pipeline. (2023).
· Anton-Păduraru, D. T., et al. (2024). Diagnosing Cystic Fibrosis in the 21st Century—A Comprehensive Review. Diagnostics, 14(7), 763.
· Anwar, S. (2024). Understanding CFTR mutation classes and modulators.
· Aqeel, H. (2025). Potential modifier genes for cystic fibrosis disease. ScienceDirect.
· Ayupova, G., et al. (2024). Population Characteristics of the Spectrum and Frequencies of CFTR Gene Mutations in Bashkortostan.
· Başaran, A. E., et al. (2021). Association Between Cystic Fibrosis Severity Markers and Genotype Classes in Children. Turkish Thoracic Journal.
· Baroni, D., et al. (2025). Research Advances on Cystic Fibrosis.
· Baroni, D. (2025). Unraveling the Mechanism of Action, Binding Sites, and Therapeutic Advances of CFTR Modulators: A Narrative Review. Current Issues in Molecular Biology, 47(2), 119.
· Birimberg-Schwartz, L., et al. (2023). Validating organoid-derived human intestinal monolayers for CFTR functional readouts. Cell Reports Methods, 3(3), 100364.
· Butnariu, L. I., et al. (2021). Genetic Modifying Factors of Cystic Fibrosis Phenotype. Journal of Clinical Medicine, 10(24), 5821.
· Caverly, L. J., & Riquelme, S. A. (2022). The impact of highly effective modulator therapy on cystic fibrosis.
· Chen, Q. (2021). A review of cystic fibrosis: Basic and clinical aspects. Wiley.
· Cimbalo, C. (2021). Elevated sweat chloride test: is it always cystic fibrosis?
· Conti, J., et al. (2022). Organoid Technology and Its Role for Theratyping in Cystic Fibrosis. Cells, 11(24), 4090.
· Cystic fibrosis: new challenges and perspectives beyond elexacaftor/tezacaftor/ivacaftor. (2025).
· Cystic Fibrosis Trust. (s. f.). CF Week, 9–15 June 2025.
· De Boeck, K. (2020). Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatrica.
· Despotes, K. A. (2022). Current state of CFTR modulators for treatment of Cystic Fibrosis.
· Donos, M. A., et al. (2025). Genetic Heterogeneity Correlated with Phenotypic Variability in Cystic Fibrosis. Frontiers in Pharmacology, 16, 12347024.
· Duffy, A., et al. (2024). A descriptive cohort study of pregnancy and parenthood in women with cystic fibrosis.
· Genetic therapies in cystic fibrosis. (2023).
· Goetz, D. M. (2021). Review of CFTR modulators. Pediatric Pulmonology.
· Gokdemir, Y., & Karadag, B. T. (2021). Sweat Testing and Recent Advances. Pediatrics.
· Graeber, S. Y., et al. (2021). Potential of intestinal current measurement for diagnosis and therapy monitoring in CF. Cells, 10(6), 1435.
· Green, D. M., et al. (2024). Cystic Fibrosis Foundation Evidence-Based Guideline.
· Hellenic Cystic Fibrosis Association. (2023, 7 de septiembre). 8th of September, Worldwide Cystic Fibrosis Day “Breath Unlimited – Be a life donor”.
· ICER. (2020). Cystic fibrosis overview.
· International Alliance of Patients' Organizations. (s. f.). Worldwide Cystic Fibrosis Day.
· Jaques, R. (2020). Novel therapeutic approaches for the management of cystic fibrosis.
· Jarzynka, S. (2025). Clinical disorders in cystic fibrosis that affect emergency care.
· Khan, F. N., Mason, K., Roe, A. H., & Tangpricha, V. (2021). CF and male health: Sexual and reproductive health, hypogonadism, and fertility. Journal of Clinical & Translational Endocrinology, 27, 100288.
· Kos, R., et al. (2022). Targeted exhaled breath analysis for detection of Pseudomonas aeruginosa in CF. J Breath Research, 16(1), 016011.
· Lepissier, A., et al. (2022). Inflammation biomarkers in sputum for clinical trials in CF lung disease. Respiratory Research, 23, 313.
· Lomunova, O. V., & Gershovich, P. (2023). RNA therapy for CF: mRNA delivery and antisense oligonucleotides.
· Lopes-Pacheco, M., et al. (2020). CFTR modulators: The changing face of cystic fibrosis in the era of precision medicine. Frontiers in Pharmacology, 11, 580.
· McGarry, M. E., et al. (2025). Cystic Fibrosis Newborn Screening: A Systematic Review.
· MedlinePlus Genetics. (2021). Cystic fibrosis.
· Minso, R., et al. (2020). Nasal potential difference and intestinal current measurements in CF diagnosis. ERJ Open Research, 6(3), 00329-2019.
· Moreno, R. M. G., et al. (2021). Treatment of pulmonary disease of cystic fibrosis.
· Muilwijk, D., et al. (2022). Forskolin-induced organoid swelling is associated with long-term CF disease progression. J Cystic Fibrosis, 21(6), 1013-1021.
· Nationwide Children’s Hospital. (s. f.). The genetics of cystic fibrosis.
· Nature Reviews Disease Primers. (2024). Cystic fibrosis: genetics, mechanisms, and therapeutics. Nature.
· Nguyen, A. L. V., et al. (2022). Metabolomic biomarkers to predict and diagnose cystic fibrosis: A systematic review. Metabolites, 12(6), 532.
· Nishat, S. M. H., et al. (2025). A new frontier in rare disease early diagnosis: AI applications and challenges. Diagnostics, 15(5), 512.
· Olivier, M. (2023). Real-life impact of highly effective CFTR modulator therapy. Frontiers in Pharmacology.
· Osadchuk, L. (2025). The Spectrum and Carrier Frequencies of Common Pathogenic CFTR Variants in Russian and Belarusian Populations.
· Osmosis. (s. f.). Cystic fibrosis: Causes & meaning.
· Parisi, G. F. (2022). Cystic fibrosis transmembrane conductance regulator (CFTR). JMHG.
· Parisi, G. F., et al. (2025). Cutting-Edge Advances in Cystic Fibrosis: From Gene Discovery to Targeted Therapies. Frontiers in Pharmacology, 16, 12026723.
· Patient Worthy. (2016, 6 de septiembre). Support Worldwide Cystic Fibrosis Day– TODAY!.
· Petrova, N., et al. (2021). Ethnic Differences in the Frequency of CFTR Gene Mutations. Frontiers in Genetics.
· Plasschaert, L. W. (2024). Current landscape of cystic fibrosis gene therapy. Frontiers in Pharmacology.
· Precision medicine advances in cystic fibrosis: Exploring genetic pathways for targeted therapies. (2024).
· Progress in precision medicine in cystic fibrosis: a focus on CFTR modulator therapy. (2022).
· Ramananda, Y., et al. (2024). Functional consequences of CFTR interactions in cystic fibrosis. Frontiers in Pharmacology, 15, 10970640.
· Respiralia. (s. f.). The Cystic Fibrosis Spanish Foundation asks the Congress of Deputies to approve an institutional declaration for ‘World CF Day’.
· Sadeghi, H., et al. (2025). Characterization of 223 infants with CRMS/CFSPID…. Journal of Cystic Fibrosis.
· Savant, A. P. (2025). Cystic fibrosis year in review 2024.
· Scotet, V., et al. (2020). Newborn screening for CF across the globe—Where is it…
· Scotet, V., et al. (2020). The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival, and Impact of CFTR Gene Discovery. Frontiers in Pharmacology, 11, 7348877.
· Sermet-Gaudelus, I., et al. (2021). Sweat chloride testing and nasal potential difference as in vivo biomarkers of CFTR activity. Diagnostics, 11(8), 1453.
· Sullivan, L. J. (2025). The aging patient with cystic fibrosis.
· Therrell, B. L., et al. (2024). Current Status of Newborn Bloodspot Screening Worldwide….
· Tümmler, B. (2025). Progress of personalized medicine of cystic fibrosis in the era of CFTR modulators. Frontiers in Pharmacology, 16, 12050259.
· Valladares, K. N. (2024). Effects of CFTR modulator therapies on pathogens in the CF lung.
· Varkki, S. D. (2024). CFTR Mutations and Phenotypic Correlations in People with Cystic Fibrosis from India. The Lancet Southeast Asia.
· Vonk, A. M., et al. (2020). Protocol for application, standardization and validation of the FIS assay in human colon organoids in CF. STAR Protocols, 1(2), 100019.
· Williams, J. D. A., Anderson, J. D., et al. (2021). CFTR function and clinical response to modulators parallel nasal epithelial organoid swelling. J Cystic Fibrosis, 20(6), 921-929.
· Yáñez-Montero, P., Mondéjar-López, P., et al. (2021). A multimodal approach to detect and monitor early lung disease in cystic fibrosis. Expert Review of Respiratory Medicine, 15(6), 761-772.
· Zubair, M. (2025). Sweat Testing - StatPearls.