


Del Diagnóstico a la Terapia Génica: Nuevas Perspectivas en el Síndrome de Down
José Hernández Jiménez
3/20/202555 min leer



Introducción
El síndrome de Down (SD), también conocido como trisomía 21, es una condición genética que afecta aproximadamente a 1 de cada 700 nacimientos a nivel global. Esta condición se caracteriza por la presencia de una copia extra del cromosoma 21, lo que provoca una serie de cambios en el desarrollo físico y cognitivo de las personas afectadas. Aunque la prevalencia del síndrome varía entre diferentes regiones, la Organización Mundial de la Salud (OMS) estima que su incidencia global se encuentra entre 1 por cada 1000 y 1 por cada 10,000 nacimientos. En América Latina, los estudios indican una mayor prevalencia debido a factores como el acceso limitado a diagnósticos prenatales y un mayor número de embarazos en mujeres mayores de 35 años.
El síndrome de Down no solo se asocia con discapacidad intelectual, sino también con una variedad de características físicas, como bajo tono muscular, estatura baja, y anomalías en la estructura facial, incluyendo ojos inclinados hacia arriba y un puente nasal plano. Además, las personas con SD presentan un mayor riesgo de desarrollar afecciones como problemas cardíacos, trastornos autoinmunes, apnea del sueño, epilepsia y enfermedad de Alzheimer en etapas tempranas de la vida. Los avances médicos han permitido mejorar la calidad de vida de quienes viven con esta condición, brindando un mejor manejo de las afecciones asociadas y un mayor apoyo social.
La investigación científica ha avanzado significativamente en el entendimiento de los mecanismos moleculares subyacentes al síndrome de Down. En particular, se ha identificado una región crítica del cromosoma 21 cuya duplicación parece jugar un papel central en los síntomas del síndrome. A pesar de los avances en el diagnóstico prenatal y los tratamientos médicos, todavía existen desafíos importantes en cuanto a la comprensión completa de los efectos cognitivos y físicos del síndrome. A medida que la ciencia avanza, se espera que el conocimiento sobre los factores genéticos y ambientales asociados con el síndrome de Down continúe mejorando, lo que permitirá optimizar las intervenciones y la integración social de las personas afectadas.
Factores de riesgo y factores protectores frente al desarrollo del Síndrome de Down
El síndrome de Down (SD) es una condición genética compleja cuyo desarrollo está influenciado por varios factores. Uno de los factores de riesgo más significativos es la edad materna avanzada, especialmente cuando la madre tiene más de 35 años. A medida que la mujer envejece, aumenta el riesgo de errores durante la división celular que ocurre al formar los óvulos, lo que puede llevar a un embarazo con síndrome de Down. Este riesgo se incrementa aún más en mujeres mayores de 40 años. Además, algunos estudios sugieren que la edad paterna avanzada también podría jugar un papel, aunque se necesita más investigación en este aspecto.
El nivel socioeconómico también es un factor de riesgo importante. Las mujeres de bajos recursos que tienen un acceso limitado a servicios de salud, incluyendo diagnósticos prenatales, tienen una mayor probabilidad de tener un hijo con síndrome de Down. Factores como una nutrición inadecuada, la exposición a productos químicos o la falta de educación podrían aumentar el riesgo de que se produzca una no disyunción cromosómica, que es la principal causa de SD. Además, la ocupación de la madre también puede influir. Investigaciones sugieren que la exposición a ciertos productos químicos en el lugar de trabajo, como los solventes, podría estar asociada con un mayor riesgo de SD, particularmente en mujeres que trabajan en la industria o en la preparación de alimentos.
Por otro lado, ciertos factores protectores pueden reducir el riesgo de tener un hijo con síndrome de Down. Uno de los más destacados es el consumo prolongado de anticonceptivos orales antes del embarazo. Estos anticonceptivos inhiben la ovulación, reduciendo el número de ciclos ovulatorios durante la vida reproductiva de la mujer. Al disminuir la cantidad de ovulaciones, se ha observado que este factor está asociado con una menor probabilidad de concebir un feto con trisomías, como la que causa el síndrome de Down.
Diversos estudios han respaldado esta hipótesis. En uno de ellos, realizado por Nagy y su equipo, se observó que las mujeres con un embarazo trisómico habían usado anticonceptivos orales durante un período más corto en comparación con aquellas que tuvieron embarazos sin trisomías. Además, las mujeres que usaron anticonceptivos orales por más tiempo presentaron un menor número de ciclos ovulatorios, lo que sugiere que el uso prolongado de estos anticonceptivos podría reducir el riesgo de trisomías.
Otro estudio realizado por Horányi y colaboradores también encontró que el uso prolongado de anticonceptivos orales y la reducción en el número de ovulaciones podrían ser factores protectores frente al síndrome de Down. Las mujeres que usaron anticonceptivos orales durante más tiempo y tuvieron menos ciclos ovulatorios mostraron una mayor tasa de embarazos con cariotipos fetales normales. Esto sugiere que la inhibición de la ovulación a través del uso prolongado de anticonceptivos podría desempeñar un papel protector en la prevención de trisomías como el síndrome de Down.
En resumen, el síndrome de Down es influenciado por una serie de factores de riesgo y protectores que involucran tanto elementos biológicos como ambientales. Estos hallazgos subrayan la importancia de seguir investigando cómo la interacción entre estos factores puede afectar el riesgo de esta condición genética.
El síndrome de Down fue descrito por primera vez en 1866 y es una de las alteraciones genéticas más comunes en el mundo. Es la principal causa de discapacidad intelectual de origen genético y se debe a la presencia de material genético extra en el cromosoma 21, razón por la cual también se le conoce como trisomía 21. Esta condición ocurre debido a un error en la división celular, llamado no disyunción, que puede suceder durante la formación de los óvulos o espermatozoides, o en las primeras etapas del desarrollo del embrión.
A nivel mundial, el síndrome de Down ocurre en aproximadamente 1 de cada 700 nacimientos, aunque esta frecuencia varía según la población estudiada. No se ha identificado una causa específica para su aparición, pero sí existen factores que pueden aumentar el riesgo, como antecedentes familiares de síndrome de Down, la edad materna menor de 19 años o mayor de 35 años, la edad paterna menor de 19 años y el sobrepeso materno antes del embarazo. Además, algunos estudios sugieren que la exposición a ciertos factores ambientales, como el consumo de tabaco y alcohol antes del embarazo, podrían influir en la aparición de la trisomía 21. Incluso, investigaciones experimentales han señalado que sustancias como el bisfenol A podrían afectar la calidad de los óvulos y espermatozoides a lo largo de varias generaciones.
El síndrome de Down puede originarse de tres maneras principales:
Trisomía 21 completa: Representa el 95% de los casos y ocurre cuando todas las células del cuerpo tienen una copia extra del cromosoma 21.
Mosaicismo: En este caso, solo algunas células tienen la trisomía 21, mientras que otras tienen una cantidad normal de cromosomas.
Translocación: Se produce cuando una parte extra del cromosoma 21 se adhiere a otro cromosoma. A diferencia de los otros dos mecanismos, la translocación puede ser hereditaria si uno de los padres es portador de una alteración cromosómica balanceada, generalmente entre el cromosoma 21 y otro cromosoma específico.
En la mayoría de los casos, la trisomía 21 completa y el mosaicismo ocurren por errores espontáneos en la división celular y no son hereditarios. Sin embargo, cuando el síndrome de Down es causado por una translocación, es posible que haya antecedentes familiares de la condición. En estos casos, se recomienda realizar estudios genéticos para evaluar el riesgo en futuras gestaciones.
La prevalencia del síndrome de Down varía entre diferentes países e incluso dentro de distintas regiones de un mismo país. Sin embargo, según la Organización Mundial de la Salud (OMS), la incidencia global generalmente se encuentra entre 1 por cada 1000 y 1 por cada 10,000 nacimientos vivos. En América Latina, el Estudio Colaborativo Latinoamericano de Malformaciones Congénitas reporta que nacen 18 niños con síndrome de Down por cada 10,000 nacidos vivos. Esto sugiere que los países en desarrollo tienen una mayor prevalencia de síndrome de Down al nacimiento, lo que podría estar relacionado con un acceso limitado a diagnósticos prenatales y un aumento en el número de embarazos en mujeres de 35 años o más.
El síndrome de Down: De la genética a la prevalencia en diversas regiones


Imagen: ctduca.com
Diagnóstico, cribado y prevención
Cribado prenatal
En países desarrollados, el cribado prenatal del síndrome de Down (SD) se ofrece como parte de la atención médica rutinaria durante el embarazo. Este proceso permite identificar embarazos con alto riesgo de SD, reduciendo así la necesidad de pruebas invasivas que podrían causar un aborto involuntario. Desde los años 80, el método principal de cribado ha combinado el análisis de ciertas sustancias en la sangre materna con la medición del grosor de la translucencia nucal (TN) del feto mediante ultrasonido en el primer trimestre. Inicialmente, se midieron los niveles de α-fetoproteína en la sangre materna y el líquido amniótico, y valores aproximadamente un 70% inferiores a los normales se asociaban con un mayor riesgo de SD. Con el tiempo, se incorporaron otros marcadores bioquímicos como la β-gonadotropina coriónica humana, el estriol no conjugado, la inhibina A y la proteína plasmática A asociada al embarazo. Dado que los niveles de estos marcadores varían con la edad gestacional, la ecografía es fundamental para una interpretación precisa de los resultados. Mediante un algoritmo computacional, se calcula el riesgo de SD en cada embarazo considerando estos valores, la edad gestacional y factores como la edad materna, antecedentes familiares, tabaquismo y diabetes. Si el resultado indica un alto riesgo, se ofrece asesoramiento genético y pruebas de diagnóstico, como la amniocentesis o la biopsia de vellosidades coriónicas.
Desde finales de los años 80, la ecografía prenatal se ha incorporado a la rutina médica para detectar posibles indicios del SD. Aunque ninguna característica anatómica es diagnóstica por sí sola, algunas señales pueden sugerir la presencia del síndrome. En el primer trimestre, una TN aumentada, la ausencia del hueso nasal o alteraciones en el flujo sanguíneo del ductus venoso pueden ser indicios. En el segundo trimestre, se realiza una ecografía detallada (entre las semanas 18 y 20), en la que se pueden observar marcadores como pliegue nucal engrosado, alteraciones en la longitud del fémur y el húmero, defectos cardíacos o intestino ecogénico. Sin embargo, muchos fetos con SD pueden presentar una ecografía aparentemente normal.
El diagnóstico prenatal del síndrome de Down (SD) ha avanzado considerablemente gracias a las técnicas de ultrasonido, que ahora permiten la detección de esta condición y de las anomalías cardíacas asociadas (CHD). En países con acceso a tecnologías de salud avanzadas, se recomienda realizar un ultrasonido detallado entre las semanas 18 y 22 del embarazo, que incluya vistas del corazón fetal. Estos estudios pueden detectar con alta precisión las anomalías cardíacas en fetos, lo que disminuye la necesidad de ecocardiogramas fetales, aunque la tasa de detección puede variar dependiendo del tipo de defecto y la experiencia del operador. En algunos casos, si el ultrasonido fetal sugiere posibles problemas cardiovasculares o si la madre presenta condiciones de riesgo, como diabetes gestacional o antecedentes familiares de CHD, se puede realizar una ecocardiografía fetal más detallada. Este examen tiene una tasa de detección superior al 90% para casos complejos de CHD.
El diagnóstico genético prenatal ha experimentado avances significativos, especialmente con la introducción de la hibridación fluorescente in situ y las pruebas no invasivas que emplean sangre materna y ADN libre de células fetales. Estas pruebas tienen una tasa de detección de hasta el 99,5% para la trisomía 21, que causa el síndrome de Down. Aunque las técnicas invasivas, como la amniocentesis o la muestra de vellosidades coriónicas, todavía se emplean en ciertos casos de alto riesgo, las pruebas no invasivas se han convertido en el estándar. La identificación temprana del síndrome de Down y las anomalías cardíacas asociadas puede influir en las decisiones sobre la continuidad del embarazo. Sin embargo, las prácticas varían según el país y la disponibilidad de recursos, y en algunos lugares de bajos recursos, los recursos para el diagnóstico prenatal no están disponibles, lo que hace esencial el diagnóstico neonatal.
Tras el nacimiento, el diagnóstico clínico de la trisomía 21 se confirma mediante pruebas genéticas rápidas, como la hibridación fluorescente in situ, que proporciona resultados en pocos días, seguidos de un cariotipo completo en 1 o 2 semanas. Todos los recién nacidos con diagnóstico de síndrome de Down deben ser evaluados para detectar signos de CHD y se recomienda realizar un ecocardiograma. En lugares donde no se dispone fácilmente de ecocardiografía neonatal, la combinación de un examen físico, electrocardiograma (ECG) y radiografía de tórax puede ayudar a identificar a los infantes que necesitan más estudios. Es importante realizar un diagnóstico temprano de estas condiciones para planificar un manejo adecuado.
Los recién nacidos con síndrome de Down enfrentan una serie de desafíos médicos, como un peso al nacer menor y una circunferencia de cabeza más pequeña, lo que aumenta su riesgo de mortalidad, siendo aproximadamente un 7,5% de los bebés con SD los que fallecen durante el periodo neonatal. Algunos defectos cardíacos, como la estenosis de las venas pulmonares o los defectos obstructivos en el lado izquierdo del corazón, también pueden aumentar este riesgo. Sin embargo, estudios han mostrado que la presencia de CHD no siempre se asocia con una mayor mortalidad hospitalaria. Además, los neonatos con síndrome de Down son más propensos a requerir ventilación mecánica o incluso oxigenación por membrana extracorpórea (ECMO), un tratamiento que se utiliza en un pequeño porcentaje de casos. La identificación y manejo temprano de estas condiciones son cruciales para mejorar los resultados en la salud de estos infantes.
Avances en la Detección Prenatal del Síndrome de Down: Integración de Aprendizaje Automático y Biomarcadores
Los modelos de aprendizaje automático (ML) están transformando la detección y el diagnóstico del síndrome de Down (SD), especialmente en el ámbito prenatal. Estas técnicas permiten mejorar las tasas de detección mientras reducen los falsos positivos. Diversos estudios han demostrado que algoritmos como los modelos de bosque aleatorio pueden alcanzar tasas de detección del 85.2% con solo un 5% de falsos positivos, superando significativamente los métodos tradicionales de laboratorio. Además, enfoques como las máquinas de vectores de soporte (SVM) y algoritmos de clasificación avanzados han mostrado resultados prometedores. Métodos de aprendizaje profundo (DL), como las redes neuronales convolucionales aplicadas a imágenes genéticas, han permitido avances en la predicción temprana del SD. Investigaciones recientes han utilizado inteligencia artificial para identificar variaciones genéticas específicas y mejorar la detección en distintos trimestres del embarazo. Asimismo, modelos basados en redes neuronales densas han demostrado una mayor precisión en la interpretación de imágenes de ultrasonido, lo que abre la puerta a diagnósticos más rápidos y confiables.
Un estudio de He et al. aplicó un modelo de bosque aleatorio a datos de 58,972 mujeres embarazadas, logrando una detección del SD del 66.7% en el conjunto inicial de datos y del 85.2% en una validación externa con 27,170 mujeres. Este resultado demuestra la superioridad de los modelos ML frente a los métodos tradicionales en la predicción prenatal. Otro estudio, realizado por Xu et al., evaluó la combinación de marcadores de ultrasonido con pruebas no invasivas (NIPT) en 856 embarazos de alto riesgo, encontrando una mayor incidencia de anomalías cromosómicas en fetos con múltiples marcadores ultrasonográficos positivos. Esta combinación de técnicas alcanzó una precisión diagnóstica del 98.29%, lo que resalta la importancia de integrar diversos métodos para mejorar la detección del SD y reducir la necesidad de pruebas invasivas como la amniocentesis.
El uso de biomarcadores en ultrasonido también ha sido optimizado mediante ML. Marcadores clave como la translucencia nucal (NT) y la presencia del hueso nasal han demostrado ser fundamentales para la evaluación del riesgo de SD. Por ejemplo, la NT sola permite detectar hasta el 70% de los casos con un 5% de falsos positivos, pero al combinarla con biomarcadores en suero materno, como la proteína plasmática A asociada al embarazo (PAPP-A) y la fracción libre de la gonadotropina coriónica humana (βhCG), la tasa de detección aumenta al 87%. Estudios recientes han desarrollado modelos predictivos avanzados basados en ML que combinan múltiples biomarcadores, lo que permite alcanzar tasas de detección cercanas al 90%. Estas mejoras en precisión han reducido la necesidad de pruebas invasivas y han permitido la implementación de estrategias de detección más personalizadas.
A nivel genético y epigenético, el aprendizaje automático también ha aportado avances importantes en la identificación de biomarcadores del SD. La trisomía del cromosoma 21 provoca alteraciones en la expresión génica que pueden ser detectadas mediante técnicas de secuenciación y análisis computacional avanzado. Investigaciones han demostrado que la expresión anormal de ciertos genes, como el del precursor de la proteína amiloide, está asociada con patologías neurodegenerativas en individuos con SD. Además, variantes genéticas como el alelo ApoE ε4 han sido identificadas como factores de riesgo para el deterioro cognitivo en esta población. A nivel epigenético, modificaciones en la metilación del ADN también han sido vinculadas con el SD. Un estudio reciente analizó perfiles de metilación en muestras de sangre neonatal y encontró regiones específicas del genoma con alteraciones epigenéticas características de la trisomía 21.
La integración de algoritmos de ML con biomarcadores genéticos y epigenéticos ha mejorado la capacidad de predicción del SD. Técnicas de minería de datos han permitido encontrar correlaciones entre la expresión de ciertos genes y la manifestación del trastorno, lo que ha llevado al desarrollo de herramientas diagnósticas más precisas. Un estudio empleó redes neuronales artificiales para analizar la expresión de genes en células amnióticas, logrando una precisión del 97% en la clasificación de casos con trisomía 21. Estas metodologías no solo optimizan la detección temprana, sino que también proporcionan información clave sobre los mecanismos moleculares involucrados en el SD, lo que podría abrir la puerta a futuras estrategias terapéuticas.
Finalmente, la integración del aprendizaje automático con biomarcadores a lo largo de los distintos trimestres del embarazo ha permitido mejorar significativamente la precisión de los modelos de detección del SD. En el primer trimestre, la combinación de NT y marcadores séricos alcanza una tasa de detección del 85%, mientras que en el segundo trimestre, la incorporación de inhibina-A y gonadotropina coriónica total mejora aún más la capacidad predictiva. Modelos de ML, como los basados en bosques aleatorios y redes neuronales profundas, han demostrado ser altamente eficaces en la identificación de patrones complejos en grandes volúmenes de datos clínicos. Estudios han mostrado que estos enfoques permiten reducir los falsos positivos y mejorar la detección de casos en poblaciones con baja incidencia de SD. En conjunto, estos avances representan un cambio de paradigma en la evaluación prenatal del SD, permitiendo diagnósticos más tempranos, precisos y accesibles para la población.
La integración de modelos de aprendizaje automático (ML) con biomarcadores ha demostrado ser una estrategia prometedora para mejorar la detección del síndrome de Down en etapas prenatales, reduciendo la tasa de falsos positivos. Estudios recientes han evidenciado que técnicas avanzadas, como los algoritmos de bosques aleatorios y redes neuronales convolucionales, logran tasas de detección superiores al 85% al combinar marcadores séricos maternos, medidas de translucencia nucal y análisis de imágenes de ultrasonido. Además, la combinación del test prenatal no invasivo (NIPT) con marcadores ultrasónicos ha mostrado una alta sensibilidad y especificidad, lo que refuerza la utilidad de enfoques multimodales en la identificación temprana de anomalías fetales. Estos avances no solo mejoran la precisión de los métodos de tamizaje prenatal, sino que también promueven un enfoque personalizado en la atención materno-fetal, optimizando la toma de decisiones médicas.
El uso de biomarcadores genéticos y epigenéticos en combinación con modelos de ML ha permitido un mejor entendimiento de los mecanismos subyacentes del síndrome de Down. Investigaciones recientes han identificado patrones de metilación del ADN asociados con la trisomía 21, lo que sugiere que los cambios epigenéticos pueden ser utilizados como indicadores tempranos de la condición. Asimismo, el análisis de la expresión génica en células amnióticas ha demostrado su viabilidad para la detección prenatal, logrando altas tasas de precisión al integrar múltiples firmas genéticas en modelos predictivos. Estos hallazgos resaltan el potencial de las herramientas computacionales para mejorar el diagnóstico y permitir estrategias de intervención más efectivas, brindando una comprensión más profunda de la biología del síndrome de Down y sus comorbilidades.
El análisis conjunto de biomarcadores a lo largo del embarazo ha optimizado la capacidad predictiva de los modelos de ML, aumentando la precisión en la identificación de embarazos de alto riesgo. En el primer trimestre, los biomarcadores tradicionales, como la translucencia nucal y los marcadores séricos maternos, han alcanzado tasas de detección cercanas al 85%. En el segundo trimestre, la inclusión de nuevos marcadores, como la gonadotropina coriónica total y la inhibina-A, ha permitido mejorar aún más la eficacia del tamizaje. Modelos basados en ML, como máquinas de vectores de soporte y bosques aleatorios, han demostrado un rendimiento superior en comparación con los métodos estadísticos convencionales. La integración de datos de distintos trimestres, junto con estrategias avanzadas para manejar desequilibrios en las bases de datos, ha fortalecido la precisión de las predicciones y ampliado su aplicabilidad clínica.
A pesar de estos avances, existen desafíos que deben abordarse antes de la implementación generalizada de estos métodos en la práctica clínica. Uno de los principales obstáculos es la necesidad de grandes conjuntos de datos representativos de diversas poblaciones para garantizar la precisión y generalización de los modelos predictivos. La variabilidad en los biomarcadores entre diferentes grupos étnicos y geográficos puede afectar la eficacia de los algoritmos, lo que subraya la importancia de realizar validaciones en poblaciones heterogéneas. Además, la integración de estas tecnologías en los sistemas de salud requiere capacitación especializada para los profesionales médicos y estrategias claras para la gestión ética de los datos, garantizando la privacidad y el consentimiento informado de los pacientes. Con investigaciones continuas y una implementación cuidadosa, la combinación de ML y biomarcadores tiene el potencial de transformar el diagnóstico prenatal del síndrome de Down, proporcionando una herramienta poderosa para mejorar la atención materno-fetal.
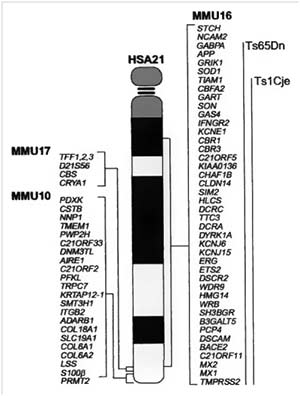
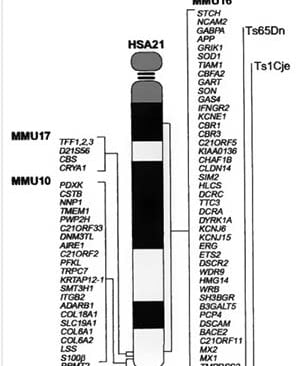
Imagen: downciclopedia.org
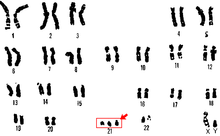
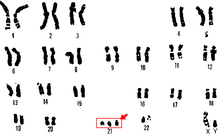
Imagen de un cariotipo donde se ve los tres cromosomas 21
Avances en cribado no invasivo
Dado que los métodos tradicionales tienen tasas de predicción limitadas (con valores predictivos positivos de solo 3-5%), se ha buscado mejorar la precisión del cribado. Desde 2011, el análisis de ADN fetal libre en la sangre materna (NIPS, por sus siglas en inglés) ha revolucionado la detección de aneuploidías como el SD. Mediante secuenciación masiva, se identifican fragmentos de ADN fetal en la sangre de la madre y se determina si hay una cantidad excesiva de material genético del cromosoma 21. Esta prueba es altamente precisa, con tasas predictivas positivas del 91% en embarazos de alto riesgo y del 82% en embarazos de bajo riesgo. Actualmente, en EE.UU., el NIPS se ofrece a mujeres con alto riesgo, mientras que en algunos países europeos, como Bélgica y los Países Bajos, ya se ofrece como prueba de cribado primaria para todas las embarazadas.
Dado que el NIPS es no invasivo y muy preciso, se han planteado interrogantes sobre su impacto en las tasas de nacimientos de bebés con SD. Aunque un estudio preliminar no encontró diferencias en los nacimientos antes y después de su implementación, aún se requieren investigaciones más amplias. Además de su utilidad para la toma de decisiones reproductivas, conocer el diagnóstico prenatal de SD puede ofrecer beneficios como una mejor preparación psicológica para los padres, la posibilidad de consultas con especialistas pediátricos antes del parto y la planificación del lugar de nacimiento para garantizar la atención adecuada al recién nacido. En el futuro, un cribado prenatal preciso podría incluso permitir intervenciones tempranas para mejorar el desarrollo cognitivo del feto.
Patologías y Síndrome Down
Cardiopatías
El diagnóstico prenatal del síndrome de Down (SD) ha avanzado considerablemente gracias a las técnicas de ultrasonido, que ahora permiten la detección de esta condición y de las anomalías cardíacas asociadas (CHD). En países con acceso a tecnologías de salud avanzadas, se recomienda realizar un ultrasonido detallado entre las semanas 18 y 22 del embarazo, que incluya vistas del corazón fetal. Estos estudios pueden detectar con alta precisión las anomalías cardíacas en fetos, lo que disminuye la necesidad de ecocardiogramas fetales, aunque la tasa de detección puede variar dependiendo del tipo de defecto y la experiencia del operador. En algunos casos, si el ultrasonido fetal sugiere posibles problemas cardiovasculares o si la madre presenta condiciones de riesgo, como diabetes gestacional o antecedentes familiares de CHD, se puede realizar una ecocardiografía fetal más detallada. Este examen tiene una tasa de detección superior al 90% para casos complejos de CHD.
El diagnóstico genético prenatal ha experimentado avances significativos, especialmente con la introducción de la hibridación fluorescente in situ y las pruebas no invasivas que emplean sangre materna y ADN libre de células fetales. Estas pruebas tienen una tasa de detección de hasta el 99,5% para la trisomía 21, que causa el síndrome de Down. Aunque las técnicas invasivas, como la amniocentesis o la muestra de vellosidades coriónicas, todavía se emplean en ciertos casos de alto riesgo, las pruebas no invasivas se han convertido en el estándar. La identificación temprana del síndrome de Down y las anomalías cardíacas asociadas puede influir en las decisiones sobre la continuidad del embarazo. Sin embargo, las prácticas varían según el país y la disponibilidad de recursos, y en algunos lugares de bajos recursos, los recursos para el diagnóstico prenatal no están disponibles, lo que hace esencial el diagnóstico neonatal.
Tras el nacimiento, el diagnóstico clínico de la trisomía 21 se confirma mediante pruebas genéticas rápidas, como la hibridación fluorescente in situ, que proporciona resultados en pocos días, seguidos de un cariotipo completo en 1 o 2 semanas. Todos los recién nacidos con diagnóstico de síndrome de Down deben ser evaluados para detectar signos de CHD y se recomienda realizar un ecocardiograma. En lugares donde no se dispone fácilmente de ecocardiografía neonatal, la combinación de un examen físico, electrocardiograma (ECG) y radiografía de tórax puede ayudar a identificar a los infantes que necesitan más estudios. Es importante realizar un diagnóstico temprano de estas condiciones para planificar un manejo adecuado.
Los recién nacidos con síndrome de Down enfrentan una serie de desafíos médicos, como un peso al nacer menor y una circunferencia de cabeza más pequeña, lo que aumenta su riesgo de mortalidad, siendo aproximadamente un 7,5% de los bebés con SD los que fallecen durante el periodo neonatal. Algunos defectos cardíacos, como la estenosis de las venas pulmonares o los defectos obstructivos en el lado izquierdo del corazón, también pueden aumentar este riesgo. Sin embargo, estudios han mostrado que la presencia de CHD no siempre se asocia con una mayor mortalidad hospitalaria. Además, los neonatos con síndrome de Down son más propensos a requerir ventilación mecánica o incluso oxigenación por membrana extracorpórea (ECMO), un tratamiento que se utiliza en un pequeño porcentaje de casos. La identificación y manejo temprano de estas condiciones son cruciales para mejorar los resultados en la salud de estos infantes.
Factores Genéticos y Moleculares en el Desarrollo de Defectos Cardíacos en el Síndrome de Down
En el contexto del síndrome de Down (DS), la trisomía 21 (T21) es la anomalía genética más comúnmente asociada con defectos cardíacos, lo que ha llevado a una extensa investigación sobre cómo los genes en el cromosoma 21 (Hsa21) contribuyen al desarrollo del corazón. Varios estudios han sugerido que los genes mapeados a Hsa21 desempeñan un papel importante en la formación cardíaca, ya que la trisomía de estos genes podría amplificar los efectos de mutaciones patogénicas o polimorfismos en otros genes relacionados con la morfogénesis del corazón. Por ejemplo, en fetos con DS, se ha demostrado que muchos de estos genes están sobrerexpresados en el corazón en desarrollo debido a la trisomía. Esta sobrerexpresión puede alterar el proceso normal de formación de las estructuras cardíacas, contribuyendo a defectos en el corazón, como la malformación de las válvulas y el tabique cardíaco.
Uno de los genes más estudiados en relación con los defectos cardíacos en el síndrome de Down es el DSCAM, que pertenece a la superfamilia de inmunoglobulinas de moléculas de adhesión celular (Ig-CAMs). Durante el desarrollo cardíaco, el DSCAM se expresa antes de la fusión de los cojines endocárdicos, y su sobreexpresión, causada por la trisomía, resulta en un aumento de la adhesión celular, lo que puede alterar la formación de los cojines cardíacos, estructuras clave en la formación de las válvulas cardíacas. Este fenómeno también se ha relacionado con otros componentes como el colágeno tipo VI, que tiene un papel importante en la formación de estructuras del corazón. En fetos con T21, se ha observado que el colágeno VI está presente en los cojines atrioventriculares (AV), y su mayor expresión podría interferir en el desarrollo adecuado de estas estructuras.
Además, existen otros genes mapeados en Hsa21 que podrían contribuir a los defectos cardíacos en el síndrome de Down. Entre ellos, el gen DYRK1A y el gen RCAN1 (también conocido como DSCR1) son relevantes, ya que ambos están involucrados en la regulación de una vía de señalización llamada NFATc, que juega un papel crucial en la morfogénesis del corazón. Estos dos genes actúan sinérgicamente para disminuir la actividad de los factores de transcripción de la familia NFATc, lo que puede alterar la formación de las válvulas y el tabique del corazón. Se ha encontrado que en fetos con T21, la expresión de DYRK1A está aumentada, mientras que la de NFATc3 está disminuida, sugiriendo que esta alteración en la regulación genética podría ser un factor clave en la patogénesis de los defectos cardíacos en el síndrome de Down.
Adicionalmente, la interacción entre genes como DYRK1A y RCAN1 con otros genes como SYNJ1, que también se encuentra en el cromosoma 21, podría tener un impacto importante en el desarrollo cardíaco. El gen SYNJ1 codifica una enzima que regula el tráfico endosomal, un proceso fundamental para la función celular y, en particular, para el transporte de proteínas dentro de las células cardíacas. Se ha demostrado que el transporte endosomal es crucial para la función del corazón, ya que controla la localización y renovación de proteínas cardíacas clave, como los canales de pacemaker y los canales de sodio. En el contexto de DS, se ha observado que los defectos en el tráfico endosomal pueden contribuir a la aparición de defectos cardíacos, lo que destaca la importancia de los procesos celulares en la patología del corazón.
En cuanto a las variantes genéticas adicionales, se ha identificado que algunas alteraciones en los genes ubicados en otros cromosomas podrían aumentar el riesgo de defectos cardíacos en individuos con síndrome de Down. Por ejemplo, variantes raras en genes como BMP4, GATA4 y CRELD1 han sido asociadas con defectos cardíacos en personas con T21, especialmente con defectos en la formación de la válvula atrioventricular (AVSD). Los estudios sugieren que estos genes, aunque no estén en el cromosoma 21, pueden actuar de manera sinérgica con la trisomía 21 para aumentar la probabilidad de defectos cardíacos. Esto resalta la complejidad genética detrás de los defectos cardíacos en el síndrome de Down, que involucra no solo la sobreexpresión de genes en Hsa21, sino también la interacción con otros genes en diferentes cromosomas.
El análisis de las variaciones en el número de copias (CNV) también ha revelado regiones en el cromosoma 21 asociadas con defectos cardíacos. Por ejemplo, se han identificado tres regiones en Hsa21 que podrían estar involucradas en el riesgo de AVSD en personas con síndrome de Down. Estas regiones incluyen genes como RIPK4 y ZBTB21, que podrían desempeñar un papel importante en la patogénesis de los defectos cardíacos. Es interesante que ZBTB21, un gen sobreexpresado en los corazones de fetos con CHD, también está involucrado en la regulación de la vía de señalización WNT/β-catenina, que es fundamental para la diferenciación cardíaca durante el desarrollo embrionario.
En paralelo a los estudios sobre los genes, también se han identificado ciertos factores no genéticos que pueden contribuir al desarrollo de defectos cardíacos en el síndrome de Down. Se ha observado que la disfunción mitocondrial es un factor relevante en la patogénesis de las enfermedades cardiovasculares, incluido el síndrome de Down. En los fibroblastos de fetos con T21, se ha encontrado que la disfunción mitocondrial es más pronunciada en aquellos con defectos cardíacos. Las mitocondrias de estos fibroblastos presentan alteraciones en su estructura, como rupturas en la membrana interna y un aumento en la producción de especies reactivas de oxígeno (ROS), lo que podría comprometer la función celular y contribuir a la aparición de defectos cardíacos. Este hallazgo sugiere que las mitocondrias podrían ser un objetivo importante en futuras investigaciones sobre los defectos cardíacos en el síndrome de Down.
Los estudios sobre los factores de transcripción como TBX20 y SRF también proporcionan información crucial sobre la formación del corazón en el síndrome de Down. El gen TBX20 juega un papel clave en la formación de las válvulas y el tabique cardíaco durante el desarrollo embrionario. Mutaciones en este gen están asociadas con una variedad de defectos cardíacos en humanos, lo que destaca su importancia en la morfogénesis cardíaca. Por su parte, SRF es un factor de transcripción fundamental en el desarrollo cardíaco tanto en embriones como en adultos. La regulación precisa de la expresión de SRF es esencial para la formación del corazón, y su alteración se ha relacionado con diversas cardiomiopatías en adultos. En fetos con T21, la expresión de SRF está disminuida, lo que podría contribuir a los defectos en el desarrollo cardíaco observados en el síndrome de Down.
En cuanto a los microARNs (miARN), varios estudios han demostrado que algunos miARNs localizados en Hsa21 están sobreexpresados en el corazón de fetos con T21, lo que podría tener un impacto negativo en el desarrollo cardíaco. El miARN-99a, let-7c y miR-155, entre otros, están involucrados en la regulación de la cardiomiogénesis y la diferenciación celular. Por ejemplo, el miR-99a inhibe la diferenciación cardíaca al regular negativamente los genes involucrados en este proceso. La sobreexpresión de miR-99a y let-7c en los fetos con T21 podría interferir en la formación normal del corazón, contribuyendo a defectos en la morfogénesis de las válvulas y el tabique.
El miR-155 es otro miARN que juega un papel crucial en la biogénesis mitocondrial, ya que regula la expresión de TFAM, un factor de transcripción esencial para la replicación y mantenimiento del ADN mitocondrial. En los corazones de fetos con T21, TFAM está subexpresado, lo que puede contribuir a la disfunción mitocondrial observada en el síndrome de Down. La alteración de la biogénesis mitocondrial es particularmente relevante para el desarrollo del corazón, ya que las mitocondrias son esenciales para la producción de energía necesaria para la contracción cardíaca.
Además de los miARNs, los estudios recientes también han comenzado a explorar el papel de los ARN no codificantes largos (lncARNs) en la patogénesis de los defectos cardíacos en el síndrome de Down. Un ejemplo destacado es el lncARN FLJ33360, que se encuentra en el cromosoma 7 y ha sido asociado con un mayor riesgo de AVSD en personas con T21. Este ARN no codificante interactúa con el gen MED10, que participa en la formación de las válvulas cardíacas en peces cebra. La identificación de este lncARN abre nuevas vías de investigación para comprender mejor los mecanismos moleculares que subyacen a los defectos cardíacos en el síndrome de Down.
En resumen, los defectos cardíacos en el síndrome de Down son el resultado de una compleja interacción entre genes localizados en el cromosoma 21 y en otros cromosomas, así como de la disfunción mitocondrial y la alteración de las vías de señalización celular. La trisomía de genes en Hsa21, como DSCAM, DYRK1A y RCAN1, junto con otros factores genéticos y epigenéticos, contribuye a la morfogénesis anormal del corazón, que se manifiesta en una amplia variedad de defectos cardíacos. La investigación futura debe seguir explorando estos mecanismos para mejorar nuestra comprensión de las causas subyacentes y las posibles estrategias terapéuticas para los defectos cardíacos en el síndrome de Down.
La regulación de la matriz extracelular (ECM, por sus siglas en inglés) juega un papel crucial en el desarrollo del sistema cardiovascular, especialmente en el contexto de enfermedades cardíacas congénitas (CHD, por sus siglas en inglés) asociadas con el síndrome de Down (DS). Durante las primeras etapas del desarrollo, la ECM es primordial, y en las fases posteriores, se experimenta un proceso de absorción y transición hacia una ECM madura, rica en colágeno y elastina. Este proceso continúa durante el período neonatal. La ECM cardíaca es una estructura dinámica y funcional que no solo proporciona soporte estructural a las células cardíacas, sino que también participa activamente en la diferenciación y desarrollo de las células cardíacas y vasculares. De hecho, los cambios en la ECM pueden desencadenar transformaciones importantes, como la transdiferenciación epitelial-mesenquimatosa (EMT), un proceso crucial en la formación de las válvulas y los septos cardíacos.
Dentro del corazón en desarrollo, las almohadillas endocárdicas desempeñan un papel esencial. Estas estructuras contienen una ECM gelatinosa que alberga células progenitoras altamente proliferativas para las válvulas y es susceptible a la remodelación continua, lo cual es fundamental para eventos morfogenéticos posteriores. Este proceso de remodelación es crítico para la formación adecuada del ventrículo y las almohadillas endocárdicas, y cualquier alteración en la regulación de la ECM puede tener efectos perjudiciales, como defectos cardíacos congénitos.
En el caso de los fetos con síndrome de Down, se ha observado que ciertos genes que codifican proteínas de la ECM están sobrerrepresentados. Un análisis de los perfiles genéticos de corazones humanos de fetos con DS reveló un aumento en la expresión de genes relacionados con proteoglicanos, colágenos y proteínas de matriz multiadherente. Esta sobreexpresión de genes que codifican componentes de la ECM podría contribuir a alteraciones en la estructura y función de la matriz, lo que a su vez puede generar anomalías cardíacas, como se observa en los defectos del septo y las válvulas cardíacas.
Además, se identificaron más de 40 genes relacionados con la ECM que están sobreexpresados en los tejidos cardíacos de fetos con síndrome de Down. Estos incluyen genes situados en el cromosoma 21, como los miembros de la familia ADAMTS, y otros genes que no están en este cromosoma pero que también están involucrados en la síntesis y remodelación de la ECM, como varios tipos de colágenos y proteínas adhesivas. Estas proteínas son esenciales para las propiedades de adhesión celular, y su alteración podría causar una mayor adherencia de las células, lo que potencialmente contribuye a defectos en el desarrollo del corazón.
En estudios con modelos murinos trans-cromosómicos que contienen un cromosoma 21 adicional, se ha demostrado que existe una migración y proliferación celular aberrantes, lo que apunta a la influencia de los genes sobre la adhesión celular. En estos modelos, las células muestran una adhesividad anormalmente alta, lo que ha sido identificado como una de las causas del fracaso en la fusión de las almohadillas endocárdicas y la persistencia de defectos como el defecto del canal atrioventricular (AVC) y las comunicación interventricular perimembranosa.
Un ejemplo clave de este fenómeno es el colágeno VI, una proteína de la ECM que tiene un papel fundamental en la anclaje de las células dentro del tejido, al interactuar con integrinas celulares y otros componentes estructurales de la matriz. En el desarrollo de las válvulas auriculoventriculares (AV), el colágeno VI y otras proteínas como el colágeno XVIII, que se expresan en las almohadillas endocárdicas, facilitan la transdiferenciación de las células endoteliales en células mesenquimatosas que forman las válvulas. Alteraciones en estos procesos de ECM pueden interferir en la formación adecuada de las válvulas cardíacas y otros defectos estructurales.
Finalmente, otros componentes de la ECM, como el versicán (VCAN), fibronectina (FN1) y fibulina (FBLN1), tienen un rol importante en la migración y proliferación celular durante el desarrollo cardíaco. Estas proteínas están involucradas en la formación de las almohadillas endocárdicas y en el proceso de EMT, que es esencial para la correcta formación de las válvulas y el tabique interventricular. Cualquier disfunción en las proteínas de la ECM puede alterar estos procesos, contribuyendo a la aparición de defectos cardíacos congénitos. Además, se ha demostrado que ciertas metaloproteinasas y proteínas de la familia ADAMTS, que participan en la degradación de la ECM, también están involucradas en la disfunción de este proceso en individuos con síndrome de Down.
Genes de la matriz extracelular y su rol en los defectos cardíacos en el síndrome de Down
El papel de RUNX1 en la regulación de los componentes de la matriz extracelular en el síndrome de Down y defectos cardíacos congénitos
RUNX1, un factor de transcripción relacionado con el gen de la trisomía 21 (Hsa21), ha sido identificado como un regulador clave en la expresión de genes relacionados con la matriz extracelular (ECM). Investigaciones han demostrado que cuando la expresión de RUNX1 se modula, los genes de la ECM son significativamente afectados. De hecho, varios genes de ECM, tanto los que se encuentran en Hsa21 como en otros cromosomas, se encuentran sobreexpresados en los corazones fetales de personas con síndrome de Down (DS). La modulación de RUNX1 a través de siRNAs reduce la expresión de varios de estos genes y mejora la capacidad migratoria de los fibroblastos trisómicos, que normalmente muestran defectos en este aspecto en comparación con controles euploides. Además, RUNX1 se encuentra en la región crítica mínima para defectos cardíacos congénitos en el síndrome de Down, lo que subraya su relevancia en estos trastornos.
RUNX1 es uno de los tres miembros de la familia RUNX de factores de transcripción, y su función principal es regular la proliferación, diferenciación y supervivencia celular. En los embriones de mamíferos, RUNX1 es el más ampliamente expresado y se encuentra en diversos tejidos, incluyendo los mesénquima del corazón y los vasos sanguíneos. Este factor es fundamental en la regulación de genes de ECM, adhesión celular y migración. Diversos estudios han mostrado que la modulación de RUNX1 influye en varias vías de señalización de integrinas, que son esenciales para la interacción de las células con la ECM. Además, se ha identificado que RUNX1 regula genes importantes para la adhesión celular, como las integrinas α y β, y varios genes relacionados con la migración celular y la ECM, incluidos los genes de las familias de ADAMTS y colágenos.
La investigación también ha revelado que RUNX1 tiene un impacto directo en los niveles de proteínas como el colágeno IV, un componente crucial de la membrana basal en el corazón. En modelos trisómicos, los fibroblastos muestran niveles elevados de colágeno IV, los cuales disminuyen cuando se atenúa la expresión de RUNX1. Este colágeno es esencial para la interacción célula-matriz, regulando procesos como la diferenciación, migración y proliferación celular. De manera similar, en estudios de cáncer, la expresión de RUNX1 se ha correlacionado con el aumento de la expresión de las metaloproteinasas de matriz (MMP), que también tienen un papel crucial en la remodelación de la ECM y la migración celular.
Finalmente, el papel de RUNX1 en el desarrollo vascular se resalta en los modelos de ratón knockout (KO) para RUNX1, que presentan un plexo coronario subdesarrollado y vasos más pequeños en las paredes ventriculares. Estos animales también muestran defectos en la estructura del corazón, como defectos en el septum ventricular y un miocardio delgado. En el corazón neonatal, la expresión de RUNX1 es más alta que en el adulto, lo que sugiere que RUNX1 podría estar involucrado en la maduración de la ECM durante el desarrollo y en la transición de un fenotipo embrionario a uno más adulto. La regulación de RUNX1 podría influir en la metilación génica durante las primeras etapas de la vida, afectando el desarrollo cardíaco y la función de la ECM en este proceso de maduración.
La familia de factores de transcripción RUNX desempeña un papel crucial en el desarrollo y el mantenimiento de los órganos en adultos. Estos genes, conocidos como RUNX1, RUNX2 y RUNX3, son miembros de la familia CBF (Core-Binding Factor) y están involucrados en una variedad de procesos en diferentes tipos celulares y tejidos. En los vertebrados, los genes RUNX tienen una organización genómica altamente conservada y poseen dos promotores alternativos, lo que les permite regular de manera eficiente la transcripción de genes específicos. Su función principal se basa en un dominio de homología llamado Runt (RHD), que les permite interactuar con secuencias de ADN específicas, además de mediar interacciones proteína-proteína, necesarias para su localización dentro del núcleo. La formación de un complejo con CBFβ es esencial para su estabilidad y para la activación o represión de los genes diana.
El RUNX1, también conocido como AML1, se considera el principal regulador de la hematopoyesis, es decir, de la formación de las células sanguíneas durante el desarrollo embrionario, y continúa jugando un papel esencial en la homeostasis del tejido sanguíneo en la etapa postnatal. Este gen, localizado en el cromosoma 21 en los seres humanos, se triplica en personas con síndrome de Down (trisomía 21), lo que sugiere que la presencia de una copia extra de RUNX1 podría estar involucrada en algunas de las características fenotípicas de este síndrome.
Estrés Oxidativo y Disfunción Celular en el Corazón de Personas con Trisomía 21
El síndrome de Down (SD) es una condición genética caracterizada por la presencia de un cromosoma 21 adicional, lo que altera diversas funciones celulares, incluyendo aquellas relacionadas con la salud del corazón. Dentro de los múltiples mecanismos biológicos afectados en este síndrome, el estrés oxidativo y la disfunción de organelos como las mitocondrias y los lisosomas han sido señalados como factores clave en el desarrollo de anomalías cardíacas. Estas alteraciones celulares pueden influir en la estructura y función del corazón, aumentando el riesgo de enfermedades cardiovasculares desde etapas tempranas de la vida.
El estrés oxidativo ocurre cuando se produce un desequilibrio entre los radicales libres, también llamados especies reactivas de oxígeno (ROS), y los sistemas antioxidantes encargados de neutralizarlos. En el SD, se ha observado una producción excesiva de ROS, lo que genera daño en las células del corazón. Un factor importante en este proceso es la enzima superóxido dismutasa (SOD1), que se encuentra sobreexpresada en las personas con esta condición. Aunque su función normal es eliminar los radicales libres, su sobreproducción en el SD conduce a un exceso de peróxido de hidrógeno, que puede causar daño oxidativo en los tejidos cardíacos. A pesar de esto, se ha observado que las personas con SD presentan una menor incidencia de enfermedades cardiovasculares isquémicas, lo que podría estar relacionado con la sobreexpresión de la proteína RCAN1, la cual parece proteger al músculo cardíaco en condiciones de isquemia y reperfusión.
Además del estrés oxidativo, las alteraciones en las mitocondrias han sido identificadas como otro factor clave en los problemas cardíacos del SD. Las mitocondrias son los organelos encargados de suministrar energía a las células mediante la producción de ATP, y su disfunción puede comprometer el funcionamiento del corazón. En estudios con fibroblastos de personas con SD, se ha observado que las mitocondrias presentan un aumento de tamaño y una estructura anómala, lo que sugiere una deficiencia en su capacidad para generar energía de manera eficiente. Estas alteraciones mitocondriales también han sido detectadas en modelos celulares de iPSCs derivadas de individuos con SD, lo que refuerza la idea de que la disfunción mitocondrial es un problema sistémico en este síndrome.
Por otro lado, los lisosomas, que son los organelos responsables de la degradación y reciclaje de componentes celulares, también muestran anomalías en el SD. Su mal funcionamiento puede provocar la acumulación de sustancias no degradadas dentro de las células, lo que afecta su viabilidad y contribuye al deterioro de tejidos, incluido el cardíaco. Se ha propuesto que la alteración del sistema de autofagia, un mecanismo mediante el cual las células eliminan componentes dañados, podría estar implicada en estas disfunciones. Dado que en otras enfermedades lisosomales, como la enfermedad de Danon, se ha encontrado una relación con el desarrollo de miocardiopatías, es posible que un mecanismo similar esté ocurriendo en el SD, aunque aún se requieren estudios más detallados en este campo.
Comprender cómo estos procesos interconectados afectan el corazón en el SD es fundamental para el desarrollo de nuevas estrategias terapéuticas. La investigación sobre el estrés oxidativo, la función mitocondrial y la regulación lisosomal no solo es relevante para mejorar la salud cardiovascular en personas con SD, sino que también puede proporcionar información valiosa sobre el desarrollo de enfermedades cardíacas en la población general. A medida que avanza el conocimiento en este ámbito, se abren nuevas posibilidades para la intervención temprana y el diseño de tratamientos más efectivos que reduzcan el impacto de estas alteraciones en la función cardíaca.
Trastornos hematopoyéticos
Los recién nacidos con síndrome de Down (SD) tienen una predisposición a desarrollar diversos problemas hematológicos, que van desde condiciones relativamente benignas, como la neutrofilia (aumento de los glóbulos blancos) y la macrocitosis (glóbulos rojos más grandes de lo normal), hasta trastornos más graves, como los trastornos mieloproliferativos transitorios (TMD). En la mayoría de los casos, estos problemas se resuelven en los primeros meses o años de vida. Sin embargo, alrededor del 10% de los pacientes con TMD asociados al síndrome de Down progresan hacia una forma de leucemia llamada leucemia megacarioblástica aguda (AMKL, por sus siglas en inglés).
La AMKL en personas con síndrome de Down está fuertemente asociada a mutaciones adquiridas en el gen GATA1, que provocan la expresión dominante de una isoforma más corta de la proteína, conocida como GATA1s. Esta variante corta carece de una región crucial para su activación (el dominio de transactivación), lo que impide que la proteína regule de manera efectiva ciertos procesos celulares, como la represión de la expresión del gen MYC, que está relacionado con el crecimiento celular descontrolado, y la red de transcripción E2F, que promueve la proliferación celular. Como resultado, la falta de control sobre estos procesos facilita la transformación de las células hacia un estado leucémico.
En consecuencia, los niños con síndrome de Down tienen un riesgo significativamente mayor de desarrollar leucemia, mientras que tienen una frecuencia mucho menor de cánceres sólidos. Los niños con trisomía 21 tienen alrededor de 500 veces más probabilidades de desarrollar AMKL, una enfermedad relativamente rara en la población general. Además, el riesgo de leucemia linfoblástica aguda pediátrica (ALL) es de 20 a 30 veces mayor en niños con síndrome de Down. Se cree que varios genes localizados en una región crítica del cromosoma 21, conocida como la "región crítica del síndrome de Down" (DSCR), incluyendo RUNX1 y DYRK1A, juegan un papel importante en aumentar el riesgo de leucemias en estas personas.
También las mutaciones en el gen RUNX1 son responsables de trastornos familiares de plaquetas, los cuales a menudo preceden a varios tipos de leucemias mieloides. Las mutaciones somáticas y las reordenaciones cromosómicas que implican la pérdida de función de RUNX1 o alteraciones en su actividad transcripcional son comunes en el síndrome mielodisplásico y en leucemias de las líneas mieloide y linfocítica, como la leucemia mieloide aguda (LMA), la leucemia linfoblástica aguda (LLA) y la leucemia crónica mielomonocítica (LCMM). Sin embargo, en los casos de leucemia asociada con el síndrome de Down (SD), el RUNX1 no suele estar mutado. En su lugar, la sobreexpresión de RUNX1, debido a la trisomía del cromosoma 21, podría disminuir el riesgo de leucemia, según los modelos experimentales en ratones.
En modelos murinos de SD, como el modelo Ts65Dn, se ha observado una enfermedad mieloproliferativa persistente, que incluye un aumento en el tamaño de los glóbulos rojos (macrocitosis) y la producción anormal de plaquetas, pero no se desarrolla leucemia. Por otro lado, otros modelos de ratón trisómicos para el cromosoma 21, como el modelo Tc1, también muestran alteraciones hematológicas similares sin desarrollar leucemia o trastornos mieloproliferativos. Estos resultados sugieren que, aunque la trisomía de RUNX1 en el síndrome de Down podría influir en el desarrollo de enfermedades hematológicas menores, no parece ser el factor principal en la leucemia asociada a este síndrome. Esto refuerza la idea de que RUNX1, en su forma normal, juega un papel más importante en la prevención de la leucemia en lugar de ser un factor etiológico en su desarrollo.
Recientemente, se ha propuesto que una isoforma corta del gen RUNX1, conocida como RUNX1a, podría estar involucrada en la leucemia asociada al síndrome de Down. Esta isoforma carece de la región de activación transcripcional del gen completo y está elevada en las células de leucemia de pacientes con síndrome de Down y leucemia mieloide (ML-DS). Los estudios han demostrado que la sobreexpresión de RUNX1a puede desplazar a la forma completa del gen, RUNX1c, de sus sitios de unión naturales, induciendo programas oncogénicos y cooperando con la mutación Gata1s, característica de este tipo de leucemia. Estos hallazgos indican que RUNX1a, a diferencia de la isoforma completa, podría contribuir al desarrollo de leucemia en células específicas del síndrome de Down, lo que muestra una compleja interacción en la regulación de las leucemias asociadas a este síndrome.
Sin embargo, en los últimos años, algunos estudios han comenzado a explorar las posibles funciones de este factor de transcripción en otros problemas relacionados con el síndrome de Down. Por ejemplo, un estudio de Mollo et al. analizó los datos de expresión génica en ratones obtenidos al sobreexpresar genes específicos del cromosoma 21 (Hsa21) y encontró que RUNX1 podría inducir la sobreexpresión de genes relacionados con la matriz extracelular (ECM). Además, la ECM apareció consistentemente como una de las categorías más afectadas en un análisis de enriquecimiento de conjuntos de genes (GSEA), lo que sugiere que RUNX1 podría tener un papel importante en los defectos congénitos del corazón, la enfermedad de Hirschsprung y la hipertensión pulmonar asociada con el síndrome de Down.
En otro estudio, Halevy et al. analizaron células madre embrionarias derivadas del síndrome de Down y observaron que la diferenciación de células progenitoras neurales (NPCs) mostraba una mayor tasa de apoptosis (muerte celular) y alteraciones en la expresión de genes relacionados con el desarrollo del cerebro. Al modificar genéticamente RUNX1, los investigadores encontraron que la reducción de su expresión disminuía la apoptosis y mejoraba la migración neuronal, lo que sugiere que RUNX1 podría estar involucrado en la regulación de estas funciones durante el desarrollo neuronal en el síndrome de Down. Este hallazgo fue respaldado por otro estudio de Liu et al., que observó que la sobreexpresión de RUNX1 en células madre pluripotentes inducidas (iPSCs) del síndrome de Down estaba asociada con una disfunción mitocondrial y un aumento de la apoptosis, lo que sugiere que RUNX1 podría ser un regulador clave de estos procesos.
Estos descubrimientos abren nuevas vías para comprender el papel de RUNX1 en el síndrome de Down, más allá de su implicación en enfermedades hematológicas. A medida que se sigan investigando sus efectos en el desarrollo y otras alteraciones fisiopatológicas del síndrome de Down, es probable que surjan más conocimientos sobre cómo este factor de transcripción y sus variantes alternativas afectan a los diferentes aspectos del síndrome.
Síndrome de Down y Salud: Aspectos Claves a lo Largo de la Vida
Las personas con síndrome de Down (SD) presentan un conjunto único de fortalezas y desafíos que pueden variar a lo largo de su vida. Mientras que algunos pueden necesitar un alto nivel de atención médica desde el nacimiento, otros pueden experimentar pocos problemas de salud. Igualmente, algunos individuos requerirán apoyo social durante toda su vida, mientras que otros pueden vivir de forma independiente. A pesar de que los problemas de salud más comunes en personas con SD incluyen defectos cardíacos congénitos (CHDs), apnea del sueño obstructiva, enfermedades tiroideas, demencia, epilepsia, trastornos gastrointestinales, problemas de audición y visión, discapacidad intelectual, trastornos del desarrollo y enfermedades mentales, los niveles de atención médica varían dependiendo de las necesidades individuales. A lo largo de su vida, estas personas deben someterse a un seguimiento regular para detectar y tratar estos problemas de salud. Mientras que existen guías de screening para niños con SD, aún no se han establecido pautas universales para adultos con esta condición.
El manejo de la salud de los adultos con SD es más complejo debido a la falta de consenso sobre quién debe supervisar su atención. En muchos casos, los médicos de atención primaria son los responsables de la gestión, aunque en algunas regiones, como el Reino Unido, los psiquiatras especializados en discapacidad intelectual pueden tomar este rol. Esto puede generar problemas, ya que, debido a la falta de pautas claras, muchos adultos con SD no reciben las revisiones médicas regulares necesarias. En algunos casos, las intervenciones solo se realizan cuando los problemas son clínicamente evidentes, lo que puede afectar la calidad de vida de estos pacientes. Debido a la complejidad de la condición y la participación de múltiples sistemas orgánicos, la atención en individuos con SD requiere un enfoque multidisciplinario que involucre equipos médicos, de cuidado social y educativo.
El manejo perinatal de las mujeres embarazadas que llevan un feto con diagnóstico confirmado de SD requiere un monitoreo continuo. Las mujeres que están embarazadas con un feto con SD tienen un mayor riesgo de aborto espontáneo, con una tasa estimada de 30% después de las 12 semanas de gestación. A medida que la madre envejece, este riesgo aumenta. Es recomendable realizar una ecografía detallada y un ecocardiograma fetal entre las 18 y 20 semanas de gestación para evaluar posibles anomalías como obstrucción gastrointestinal, quilotórax, hidropesía fetal y restricción del crecimiento intrauterino. Si se detectan anormalidades, se debe incrementar la vigilancia fetal. Además, se debe informar al equipo pediátrico local sobre cualquier anomalía para que puedan planificar los cuidados postnatales del bebé.
Aproximadamente el 50% de las personas con SD padecen defectos cardíacos congénitos, siendo los más comunes el defecto del canal auriculoventricular (AVSD), el defecto del septo ventricular y el defecto del septo auricular. Estos problemas pueden afectar gravemente la calidad de vida del individuo. Durante el embarazo, se recomienda realizar una ecografía fetal para evaluar el corazón del bebé. Después del nacimiento, se debe llevar a cabo una evaluación cardiológica y otro ecocardiograma dentro del primer mes de vida. El tratamiento de estos defectos es el mismo que en la población general e incluye reparaciones quirúrgicas. La tasa de mortalidad tras la cirugía en niños con SD es igual o inferior a la de la población general. A lo largo de su vida, todas las personas con SD deben ser evaluadas anualmente para detectar signos de enfermedades adquiridas de las válvulas cardíacas y de insuficiencia cardíaca.
La apnea obstructiva del sueño es un problema común en personas con SD, con una prevalencia estimada de entre el 54 y el 90%. Es importante realizar un seguimiento de los síntomas de apnea del sueño, como ronquidos fuertes, respiración pesada, noches inquietas y somnolencia diurna. Otros síntomas neurocognitivos incluyen irritabilidad, depresión, paranoia y problemas de comportamiento. Se recomienda realizar una polisomnografía nocturna en todos los niños con SD antes de los 4 años, independientemente de si presentan síntomas o no. En algunos casos, el uso de oximetría en casa puede ayudar a identificar a los niños en riesgo de apnea del sueño, reduciendo la necesidad de estudios diagnósticos completos. El tratamiento incluye el uso de dispositivos de presión positiva continua en las vías respiratorias (CPAP), dispositivos de avance mandibular y pérdida de peso. En algunos casos, se pueden considerar intervenciones quirúrgicas, como la adenoidectomía y la amigdalectomía, aunque la apnea puede persistir después de la cirugía.
El hipotiroidismo congénito afecta a aproximadamente el 1% de las personas con SD, y más del 50% de los neonatos con SD presentan resultados anormales en las pruebas de función tiroidea. La presencia de enfermedades tiroideas es más frecuente con el paso de los años, y el riesgo de disfunción tiroidea autoinmune aumenta con la edad. Dado que el diagnóstico clínico de las afecciones tiroideas puede ser complicado, es esencial realizar un análisis de sangre regular. Se deben medir los niveles de hormona estimulante de la tiroides (TSH) y de tiroxina (T4) en el posparto, a los 6 meses y a los 12 meses de edad. Posteriormente, los niveles de TSH deben medirse anualmente para garantizar un monitoreo continuo de la función tiroidea.
Un gran porcentaje de personas con SD desarrollan enfermedad de Alzheimer (EA) de inicio temprano, lo que se relaciona con la sobreproducción de la proteína precursora amiloide (APP). La demencia es la causa principal de muerte en el 70% de los adultos mayores con SD. Los síntomas clínicos suelen aparecer después de los 40 años, y el 77% de los individuos con SD de entre 60 y 69 años y hasta el 100% de aquellos mayores de 70 años desarrollarán un deterioro cognitivo. Este deterioro debe evaluarse teniendo en cuenta el perfil cognitivo individual, ya que la presencia del alelo APOEε4 puede acelerar el deterioro cognitivo relacionado con la EA. Los déficits en memoria y atención son comunes en las primeras etapas de la enfermedad, al igual que en los individuos con EA esporádica, pero a menudo no se reconocen hasta que aparecen cambios en el comportamiento.
Se recomienda realizar una evaluación de referencia de la cognición y la adaptación funcional a los 30 años de edad en todas las personas con SD para ayudar a un monitoreo y diagnóstico futuros. El tratamiento debe centrarse en la detección temprana y las medidas de apoyo. El uso de inhibidores de la acetilcolinesterasa, como el donepezilo, ha demostrado ciertos beneficios, aunque algunos individuos pueden experimentar efectos adversos, como la disminución de la frecuencia cardíaca. Las personas con SD y demencia deben contar con un adecuado apoyo, ya que sus necesidades aumentarán a medida que la enfermedad progrese. El objetivo debe ser mantener a las personas en su entorno familiar, y las transferencias a otros proveedores deben basarse en las circunstancias y necesidades individuales.
En cuanto a la epilepsia, alrededor del 8% de los niños con SD presentan convulsiones, con dos picos en la edad de inicio: uno antes de los 3 años y otro después de los 30 años. La epilepsia infantil más frecuente es el síndrome de West, que se caracteriza por espasmos infantiles y patrones anormales de actividad cerebral. El tratamiento de la epilepsia depende de su causa, pero generalmente se utiliza medicación anticonvulsiva, la cual es efectiva para reducir la actividad convulsiva en la mayoría de los casos.
La pérdida de audición conductiva es común en personas con SD, especialmente debido a otitis media con efusión. Se recomienda realizar pruebas auditivas de referencia después del nacimiento y luego cada seis meses hasta que el niño comience la escuela, y al menos anualmente después. Reconocer y tratar tempranamente los problemas de audición puede reducir el riesgo de pérdida de audición a largo plazo. Con el tiempo, la pérdida auditiva sensorineural también es común en adultos. Los dispositivos de asistencia auditiva y los implantes cocleares son tratamientos eficaces. Además, el uso de terapia del habla y ayudas para la comunicación, como el lenguaje de señas, pueden ser beneficiosos.
Las personas con SD también tienen un mayor riesgo de problemas oculares, como errores de refracción, cataratas y ambliopía. Se debe realizar un examen oftalmológico al nacer y luego de forma regular a lo largo de la vida, idealmente cada 1 o 2 años. Si se detectan cataratas, es común realizar cirugía, la cual generalmente tiene buenos resultados. Además, existe el riesgo de inestabilidad atlantoaxoidea, una afección que implica la subluxación de las vértebras cervicales superiores, lo que puede aumentar el riesgo de lesiones medulares, especialmente durante actividades físicas.
Los trastornos de la salud mental son más comunes en las personas con SD que en la población general. Las personas con SD tienen una mayor prevalencia de trastornos de ansiedad y depresión. Un pequeño porcentaje de adolescentes y adultos jóvenes con SD experimentan regresión aguda, lo que implica una pérdida de habilidades y mayor dependencia. Actualmente, no se comprende completamente la causa de esta regresión, pero parece estar relacionada con situaciones de estrés emocional. El diagnóstico de problemas de salud mental en individuos con SD puede ser complicado debido a la discapacidad intelectual, dificultades de comunicación y síntomas atípicos.
La neurocognición en las personas con SD está frecuentemente afectada por una discapacidad intelectual de leve a moderada. Se sabe que las personas con SD tienen una mayor prevalencia de trastornos como el autismo y el TDAH, por lo que es importante
Hacia una Terapia Genética para el Síndrome de Down: Eliminación de Cromosomas con CRISPR
El síndrome de Down (SD) es una condición genética causada por la presencia de un cromosoma 21 extra, lo que altera la expresión génica y contribuye a diversas manifestaciones clínicas. A pesar de los avances en la comprensión de esta trisomía, aún no existen tratamientos que corrijan directamente la carga genética adicional. Sin embargo, recientes investigaciones han explorado la posibilidad de eliminar selectivamente el cromosoma extra utilizando la tecnología de edición genética CRISPR/Cas9. Este enfoque innovador no solo permitiría restaurar un perfil genético más equilibrado en las células afectadas, sino que también podría abrir nuevas vías para el desarrollo de futuras terapias para el SD. En este estudio, se analiza la aplicación de CRISPR/Cas9 para la eliminación específica del cromosoma 21 adicional, evaluando su eficacia, impacto en la expresión génica y potencial para corregir defectos celulares característicos del SD.
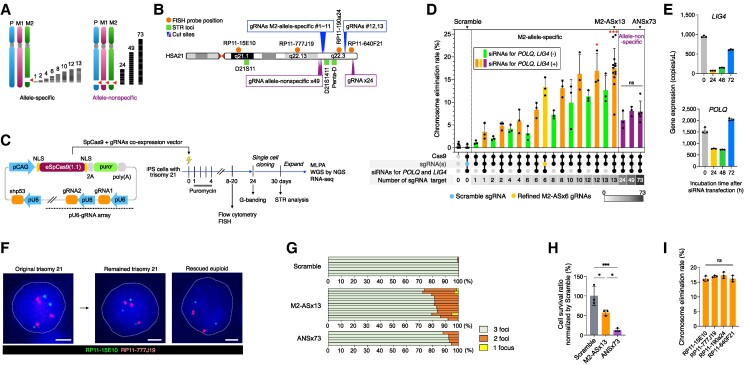
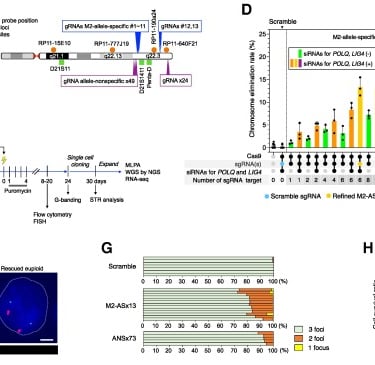
Imagen: Hashizume R, Wakita S, Sawada H, Takebayashi SI, Kitabatake Y, Miyagawa Y, Hirokawa YS, Imai H, Kurahashi H. Trisomic rescue via allele-specific multiple chromosome cleavage using CRISPR-Cas9 in trisomy 21 cells. PNAS Nexus. 2025 Feb 18;4(2):pgaf022.
En un estudio reciente, se crearon células madre pluripotentes inducidas (iPS) con trisomía 21, a partir de fibroblastos dérmicos, y se generaron tres líneas celulares con disomía inducida mediante la eliminación de cromosomas. Para llevar a cabo esta corrección cromosómica, se utilizó el sistema CRISPR/Cas9, que permite editar genes de manera precisa. Se seleccionó el alelo M2 del cromosoma 21 como objetivo para la edición genética debido a que algunos genes de este cromosoma pueden ser sensibles al origen materno. Después de diseñar guías de ARN altamente específicas, se comprobó la efectividad y especificidad del sistema mediante secuenciación genómica, asegurando que no se produjeron efectos indeseados fuera de las secuencias objetivo.
Con el fin de optimizar la corrección cromosómica, se identificaron 15,695 secuencias exclusivas del alelo M2, muchas de las cuales estaban localizadas en la región subtelomérica del cromosoma 21. A partir de estas secuencias, se seleccionaron varias candidatas para evaluar su eficiencia en la inducción de cortes cromosómicos. El análisis mostró que algunos de los cortes realizados eran altamente específicos para el alelo M2, mientras que otros afectaban también secuencias fuera de la región objetivo. Posteriormente, se probó la eliminación del cromosoma extra mediante cortes en diferentes puntos del cromosoma, y se observó que la tasa de eliminación aumentaba conforme se realizaban más cortes. Además, se mejoró el proceso de eliminación al suprimir genes clave involucrados en la reparación del ADN, lo que resultó en un incremento de la tasa de eliminación cromosómica.
Una comparación entre dos estrategias de eliminación cromosómica mostró que el enfoque dirigido a un solo alelo, el M2, resultó ser mucho más eficiente que la estrategia dirigida a los tres alelos del cromosoma 21. Esta precisión en la selección del alelo permitió mejorar la eficacia del método y reducir los efectos secundarios no deseados. Para confirmar la eliminación específica del cromosoma extra, se usaron células reporteras con marcadores fluorescentes insertados en cada alelo del cromosoma 21. La pérdida de fluorescencia en el alelo M2 tras la transfección con el vector M2-AS × 13 confirmó la eliminación exitosa del cromosoma extra. Además, los análisis de cariotipos mostraron que las células corregidas presentaban un cariotipo disómico normal, sin alteraciones estructurales.
A pesar de los avances en la eliminación cromosómica, algunos sitios aún mostraban cortes sin eliminar completamente el cromosoma extra. Esto sugiere que la reparación del ADN permitió la retención del cromosoma en ciertos casos, aunque no se observaron alteraciones genómicas importantes. Para mejorar aún más la eficiencia del proceso, se desarrolló un nuevo vector con solo seis guías de ARN, que mostró ser más eficiente en la eliminación cromosómica que el sistema original de 8 guías, aunque ligeramente menos eficiente que el sistema de 13 guías. Estos resultados destacan la importancia tanto del número de cortes como de la precisión en la selección del alelo en la mejora de la corrección cromosómica.
Además de los experimentos de corrección cromosómica, se llevó a cabo un análisis de la expresión génica en las células iPS para evaluar si la corrección del cariotipo restauraba los perfiles de expresión génica alterados en las células con trisomía 21. Los resultados mostraron que las células corregidas, en comparación con las células trisómicas originales, mostraban una mejora en la expresión génica. Específicamente, un porcentaje significativo de genes relacionados con procesos metabólicos, como la ribosa y el colesterol, mostraron una disminución en su expresión, mientras que genes asociados con el desarrollo celular y la neurogénesis, como aquellos involucrados en la formación del sistema nervioso, se expresaron a niveles más altos en las células rescatadas. Este hallazgo sugiere que la corrección cromosómica no solo restaura la cantidad de cromosomas, sino también la funcionalidad de las vías génicas afectadas.
Finalmente, se realizaron estudios adicionales para evaluar los efectos de la corrección cromosómica en las células madre pluripotentes inducidas en cuanto a su potencial de desarrollo. El análisis de enriquecimiento de conjuntos de genes mostró que las células rescatadas activaban genes relacionados con el desarrollo temprano del sistema nervioso, como la proliferación de células precursoras neuronales. Este hallazgo es relevante para el síndrome de Down, ya que los defectos en el desarrollo del sistema nervioso son una característica prominente de la enfermedad. Los resultados sugieren que la corrección cromosómica puede tener un impacto significativo en la corrección de defectos organogénicos, particularmente en el sistema nervioso, lo que podría abrir nuevas vías para el tratamiento de trastornos relacionados con la trisomía 21 y mejorar la calidad de vida de los pacientes.
Este estudio se enfoca en abordar uno de los mayores desafíos relacionados con el síndrome de Down (SD), específicamente la identificación de los genes del cromosoma 21 que son responsables de los fenotipos clínicos observados en las personas con trisomía 21. A pesar de los esfuerzos realizados, aún no existe un consenso claro sobre qué cambios específicos en la expresión génica son característicos de esta condición. Esto se debe en parte a la variabilidad genética entre individuos, lo que influye de manera significativa en los perfiles de expresión génica, haciendo difícil identificar patrones consistentes y confiables que sean exclusivos de la trisomía 21. Al comparar los datos obtenidos en este estudio con los de una investigación anterior sobre células madre pluripotentes inducidas (iPS) de gemelos monocigóticos discordantes para trisomía 21, se observó una baja concordancia entre los dos conjuntos de datos. Solo un pequeño porcentaje de los genes mostraron coincidencias en la expresión, lo que sugiere que las diferencias genéticas individuales son un factor clave en la complejidad de los perfiles de expresión génica en el síndrome de Down.
La hipótesis de los fenotipos asociados con la aneuploidía ha ganado atención recientemente, sugiriendo que la ganancia de una copia adicional de un cromosoma podría generar fenotipos, tanto genéticos como asociados a la aneuploidía, de manera independiente a los genes presentes en ese cromosoma. Este fenómeno, que se observa en organismos desde levaduras hasta células humanas, incluye características como una menor viabilidad celular, morfología nuclear anómala y un aumento en la demanda metabólica. Dado esto, una posible estrategia para abordar los problemas fenotípicos derivados de la trisomía sería inducir una "rescate de trisomía", lo cual restauraría el estado diploide mediante la eliminación de material genético adicional, una técnica que podría ser radical, pero efectiva.
A pesar de la viabilidad de esta estrategia, la eliminación total de cromosomas ha demostrado ser extremadamente difícil desde el punto de vista técnico. Se han intentado métodos como insertar genes letales o activar mecanismos de silenciamiento del cromosoma extra, pero ambos enfoques requieren modificaciones genéticas profundas y no son compatibles con el uso clínico. En cambio, este estudio propone un método más sencillo y directo que utiliza la edición genética con CRISPR/Cas9 para cortar específicamente el alelo del cromosoma objetivo y eliminarlo por completo de la célula, lo que resulta en un enfoque más práctico para la aplicación clínica.
El sistema CRISPR/Cas9 es conocido por su capacidad para inducir grandes deleciones cromosómicas, aunque las pérdidas de cromosomas completos son relativamente raras, especialmente en células somáticas no embrionarias. Sin embargo, en este estudio, se logró utilizar esta propiedad de Cas9 para inducir la eliminación selectiva de cromosomas en células con trisomía 21. Aunque la pérdida total del cromosoma en células con cariotipos normales puede ser perjudicial, la reprogramación de células trisómicas a células madre pluripotentes inducidas (iPS) facilita la eliminación dirigida de cromosomas extra, un proceso aprovechado por el método propuesto. Además, se observó que, a diferencia de los estudios previos, en células trisómicas no ocurrieron truncamientos terminales del cromosoma, sino que estos fueron eliminados en su totalidad, lo que muestra una diferencia clave en comparación con otros enfoques.
En cuanto a las modificaciones genómicas que acompañan a la eliminación cromosómica, este estudio encontró que los cortes inducidos por Cas9 provocaron alteraciones genómicas, como inserciones y deleciones pequeñas, que no fueron detectadas mediante análisis cromosómicos convencionales como el G-banding. Sin embargo, en los casos donde los cromosomas no fueron eliminados completamente, se introdujeron mutaciones en los cromosomas residuales, lo que subraya la importancia de mejorar las tasas de eliminación cromosómica para reducir la carga genética indeseada. Para mejorar estos resultados, se utilizó una estrategia de supresión temporal de los mecanismos de reparación del ADN, lo que aumentó significativamente la tasa de eliminación cromosómica, al promover que las células no repararan los cortes, sino que eliminaran el cromosoma dañado.
Por último, se discutieron diversas técnicas de edición genética que han intentado eliminar cromosomas específicos, incluyendo enfoques con nucleasas y métodos basados en la manipulación de las proteínas del huso mitótico. Sin embargo, a pesar de sus ventajas, estas técnicas presentan limitaciones en términos de control sobre la pérdida o ganancia de cromosomas. En comparación, la estrategia AS (Alelo Específico) empleada en este estudio tiene la ventaja de enfocarse solo en un cromosoma específico, evitando efectos no deseados como la disomía uniparental, lo que la hace más eficaz en comparación con otros métodos. A pesar de su potencial, este enfoque requiere más optimización, especialmente para mejorar su aplicabilidad clínica.
En resumen, este estudio demuestra que es posible corregir la anomalía cromosómica en células con trisomía 21 mediante la eliminación selectiva del cromosoma 21 extra usando CRISPR/Cas9, una estrategia que tiene el potencial de convertirse en una intervención terapéutica para el síndrome de Down. Los resultados también muestran que la supresión temporal de los genes involucrados en la reparación del ADN mejora la tasa de eliminación de cromosomas, lo que sugiere que este enfoque podría restaurar tanto la expresión génica como los fenotipos celulares a niveles normales. Aunque este método ha demostrado ser efectivo en células madre pluripotentes y células diferenciadas, aún se necesita más investigación para optimizar la tasa de eliminación cromosómica, evaluar la seguridad y establecer sistemas de entrega in vivo para su aplicación clínica.
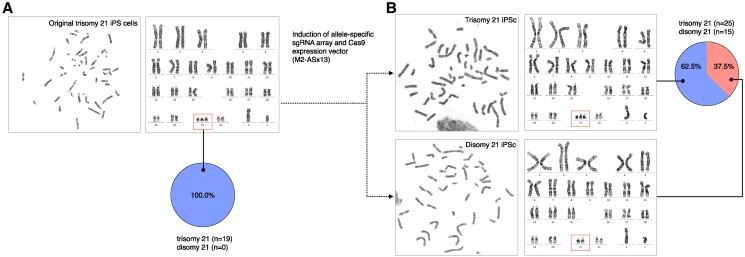
Imagen: Hashizume R, Wakita S, Sawada H, Takebayashi SI, Kitabatake Y, Miyagawa Y, Hirokawa YS, Imai H, Kurahashi H. Trisomic rescue via allele-specific multiple chromosome cleavage using CRISPR-Cas9 in trisomy 21 cells. PNAS Nexus. 2025 Feb 18;4(2):pgaf022.
Referencias:
Hashizume R, Wakita S, Sawada H, Takebayashi SI, Kitabatake Y, Miyagawa Y, Hirokawa YS, Imai H, Kurahashi H. Trisomic rescue via allele-specific multiple chromosome cleavage using CRISPR-Cas9 in trisomy 21 cells. PNAS Nexus. 2025 Feb 18;4(2):pgaf022.
Dohl J, Treadwell Z, Norris C, Head E. Calcineurin inhibition may prevent Alzheimer disease in people with Down syndrome. Alzheimers Dement. 2025 Mar;21(3):e70034.
Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, Sherman SL, Reeves RH. Down syndrome. Nat Rev Dis Primers. 2020 Feb 6;6(1):9.
Blanco-Montaño A, Ramos-Arenas M, Yerena-Echevarría BA, Miranda-Santizo LD, Ríos-Celis AL, Dorantes-Gómez AT, Morato-Rangel AJ, Meza-Hernández JA, Acosta-Saldívar ED, Aguilar-Castillo CD, Cárdenas-Conejo A. Factores de riesgo en el origen del síndrome de Down [Risk factors in the origin of Down syndrome]. Rev Med Inst Mex Seguro Soc. 2023 Sep 4;61(5):638-644.
Yahia S, Metawea M, Megahed A, ELshawaf W, Wahba Y, Mahmoud R. The prevalence of hearing impairment in infants and children with down syndrome a cross sectional study in a Tertiary Care Center. Sci Rep. 2025 Mar 4;15(1):7570.
Danaei M, Rashnavadi H, Yeganegi M, Dastgheib SA, Bahrami R, Azizi S, Jayervand F, Masoudi A, Shahbazi A, Shiri A, Aghili K, Mazaheri M, Neamatzadeh H. Advancements in machine learning and biomarker integration for prenatal Down syndrome screening. Turk J Obstet Gynecol. 2025 Mar 10;22(1):75-82.
Rozen EJ, Ozeroff CD, Allen MA. RUN(X) out of blood: emerging RUNX1 functions beyond hematopoiesis and links to Down syndrome. Hum Genomics. 2023 Sep 5;17(1):83.
Mollo N, Scognamiglio R, Conti A, Paladino S, Nitsch L, Izzo A. Genetics and Molecular Basis of Congenital Heart Defects in Down Syndrome: Role of Extracellular Matrix Regulation. Int J Mol Sci. 2023 Feb 2;24(3):2918.
Sharaf R, Garout W, Sharaf R. Prevalence of Congenital Heart Defects in Individuals With Down Syndrome in Saudi Arabia: A Systematic Review and Meta-Analysis. Cureus. 2022 Nov 18;14(11):e31638.
Dimopoulos K, Constantine A, Clift P, Condliffe R, Moledina S, Jansen K, Inuzuka R, Veldtman GR, Cua CL, Tay ELW, Opotowsky AR, Giannakoulas G, Alonso-Gonzalez R, Cordina R, Capone G, Namuyonga J, Scott CH, D'Alto M, Gamero FJ, Chicoine B, Gu H, Limsuwan A, Majekodunmi T, Budts W, Coghlan G, Broberg CS; for Down Syndrome International (DSi). Cardiovascular Complications of Down Syndrome: Scoping Review and Expert Consensus. Circulation. 2023 Jan 31;147(5):425-441.
Venegas-Zamora L, Bravo-Acuña F, Sigcho F, Gomez W, Bustamante-Salazar J, Pedrozo Z, Parra V. New Molecular and Organelle Alterations Linked to Down Syndrome Heart Disease. Front Genet. 2022 Jan 18;12:792231.
Jones JT, Kitchen J, Talib N. Down Syndrome-Associated Arthritis (DA): Diagnostic and Management Challenges. Pediatric Health Med Ther. 2022 Mar 14;13:53-62.
Bates ML, Vasileva A, Flores LDM, Pryakhina Y, Buckman M, Tomasson MH, DeRuisseau LR. Sex differences in cardiovascular disease and dysregulation in Down syndrome. Am J Physiol Heart Circ Physiol. 2023 Apr 1;324(4):H542-H552.
Ferrari M, Stagi S. Autoimmunity and Genetic Syndromes: A Focus on Down Syndrome. Genes (Basel). 2021 Feb 13;12(2):268.
Sinton JW, Cooper DS, Wiley S. Down syndrome and the autonomic nervous system, an educational review for the anesthesiologist. Paediatr Anaesth. 2022 May;32(5):609-616.
(anuncios)