


El cáncer infantil: una perspectiva global y sus desafíos
José Hernández Jiménez
2/14/202519 min leer


El cáncer infantil: una perspectiva global y sus desafíos
Introducción
El cáncer infantil es un conjunto diverso de neoplasias malignas que abarca una variedad de enfermedades con características muy diferentes en cuanto a su aparición, causas, tratamiento, cuidados y pronóstico. A pesar de ser un grupo heterogéneo, las investigaciones y avances médicos han logrado mejorar notablemente las tasas de supervivencia en las últimas décadas, transformando la perspectiva de muchos niños afectados por esta enfermedad.
Avances en el tratamiento y la supervivencia
A nivel global, los avances en la investigación sobre el cáncer infantil han permitido una mejora significativa en los tratamientos y en los índices de supervivencia. En países desarrollados, como Estados Unidos, el 85% de los niños diagnosticados con cáncer sobreviven más de cinco años. Sin embargo, en otras partes del mundo, especialmente en regiones de África, América Latina y Asia, las tasas de supervivencia son considerablemente más bajas. Por ejemplo, en algunas áreas de África del Este, estas tasas pueden ser tan solo del 8%. Estas disparidades en la supervivencia reflejan las diferencias en el acceso a tratamientos especializados, recursos médicos y la capacitación de los profesionales de salud.
Desigualdad geográfica en las tasas de supervivencia
Las tasas de supervivencia varían no solo entre países, sino también dentro de las regiones más desarrolladas, donde no todos los tipos de cáncer infantil presentan el mismo pronóstico. El cáncer linfático agudo (ALL) tiene mejores resultados en comparación con otros tipos, como el cáncer mieloide agudo (AML). Además, enfermedades como el linfoma de Burkitt o el linfoma de Hodgkin, presentan tasas de supervivencia aún más altas. Sin embargo, las diferencias en la supervivencia no solo se deben al tipo de cáncer, sino también a factores sociales y geográficos, lo que resalta la persistente desigualdad en el acceso a la atención médica.
Factores socioeconómicos y demográficos en el pronóstico
La edad, el sexo y el estatus socioeconómico juegan un papel crucial en los resultados de los pacientes con cáncer infantil. Los neonatos y los bebés suelen tener mejores perspectivas de supervivencia en algunos tipos de cáncer. No obstante, el estatus socioeconómico de las familias sigue influyendo negativamente en los resultados, incluso en países con sistemas de salud avanzados. Esto pone de manifiesto la importancia de abordar tanto los factores sociales como los económicos para garantizar una atención equitativa para todos los niños afectados por esta enfermedad.
Efectos secundarios y calidad de vida de los supervivientes
El tratamiento del cáncer infantil no solo se centra en erradicar la enfermedad, sino también en los efectos secundarios a largo plazo que los niños pueden experimentar. El dolor persistente durante y después del tratamiento es uno de los problemas más comunes, afectando la calidad de vida de los supervivientes. Además, los niños que sobreviven al cáncer pueden enfrentar un envejecimiento prematuro y un aumento en la fragilidad física, lo que incrementa el riesgo de enfermedades crónicas, caídas y hospitalizaciones en el futuro. Este envejecimiento acelerado no es exclusivo de los supervivientes del cáncer infantil, sino que también afecta a otras personas que padecen enfermedades crónicas relacionadas con el envejecimiento.
Retos y perspectivas futuras
A pesar de los avances en el tratamiento y la mejora de las tasas de supervivencia, el cáncer infantil sigue presentando retos significativos a nivel global. Las desigualdades en el acceso a la atención médica y los efectos secundarios del tratamiento son cuestiones que aún requieren atención. La investigación continua y la colaboración internacional son fundamentales para reducir estas disparidades, mejorar la calidad de vida de los supervivientes y, en última instancia, avanzar en la cura de estas enfermedades.
Este artículo aborda los principales desafíos y avances en el tratamiento del cáncer infantil, destacando la necesidad urgente de soluciones equitativas y sostenibles en el acceso a la atención médica para todos los niños, independientemente de su lugar de residencia o situación económica.
Diagnóstico y Predisposición Genética al Cáncer Infantil
El diagnóstico de cáncer infantil con predisposición genética involucra una serie de pruebas y evaluaciones clínicas centradas en el historial familiar y las características fenotípicas del paciente. Además, la genética juega un papel crucial, ya que ciertas variantes genéticas pueden aumentar el riesgo de desarrollar cáncer a una edad temprana. En esta sección, se discuten los métodos de diagnóstico y los principales síndromes de predisposición al cáncer infantil, así como el impacto de la genética en el tratamiento.
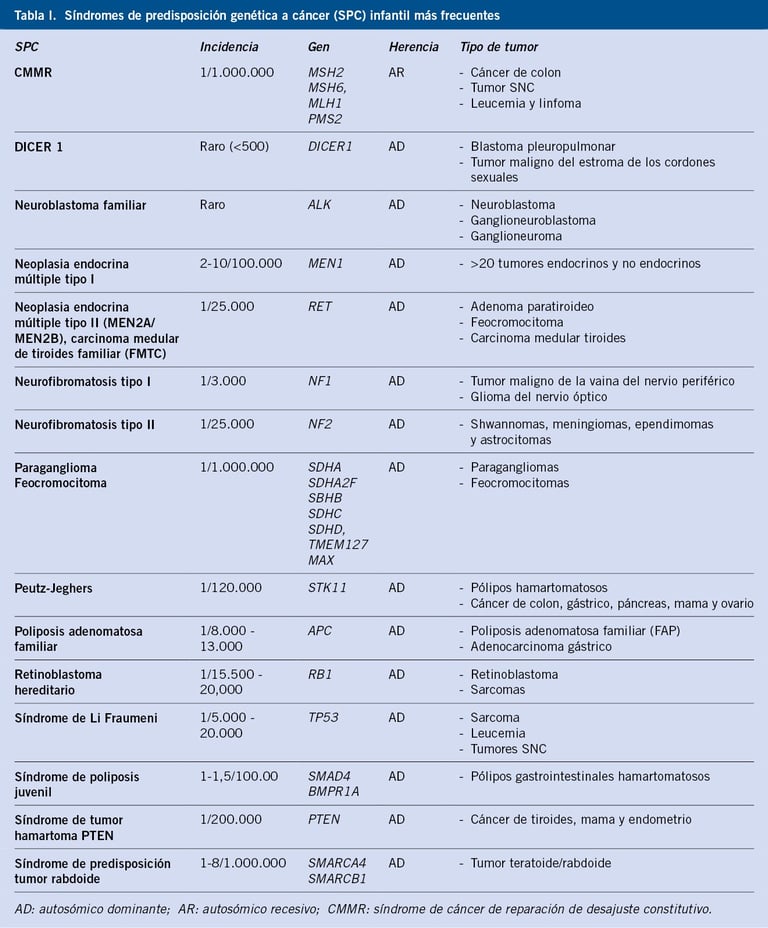
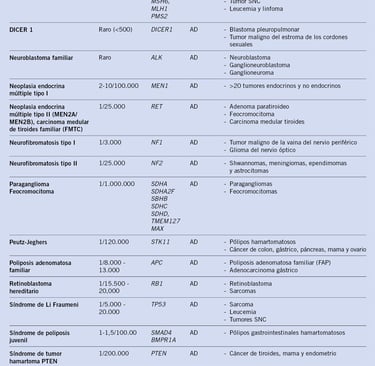
Tabla: pediatriaintegral.es
Diagnóstico Clínico y Evaluación del Historial Familiar
Tradicionalmente, el diagnóstico de los síndromes de predisposición al cáncer infantil se basa en la sospecha clínica. Los médicos suelen comenzar evaluando el historial familiar, buscando patrones que sugieran una predisposición hereditaria. Es importante que se incluya información sobre al menos tres generaciones, ya que esto puede ser indicativo de una mutación genética heredada. Sin embargo, un historial familiar negativo no necesariamente excluye el diagnóstico, ya que existen mutaciones de novo (variantes genéticas nuevas que no provienen de los padres) y otros factores como la penetrancia reducida que pueden no ser evidentes en la historia familiar.
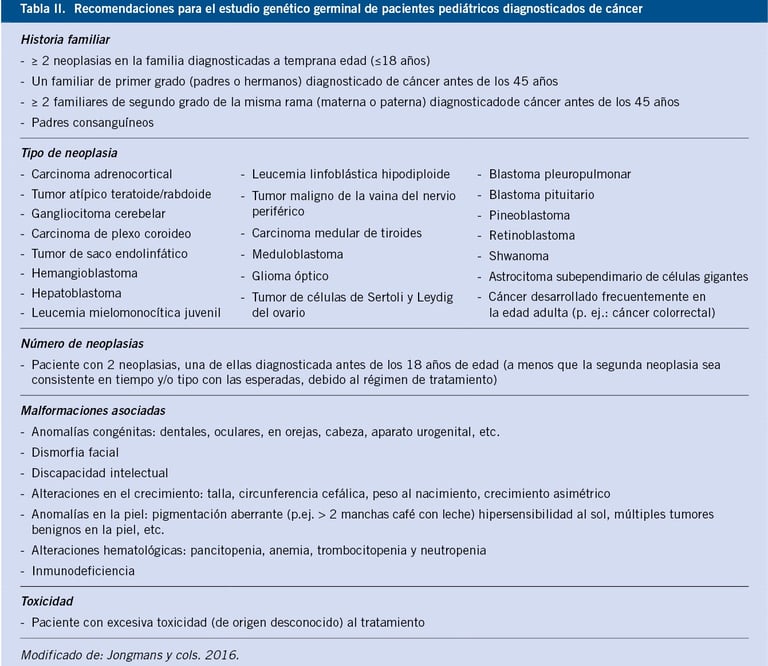
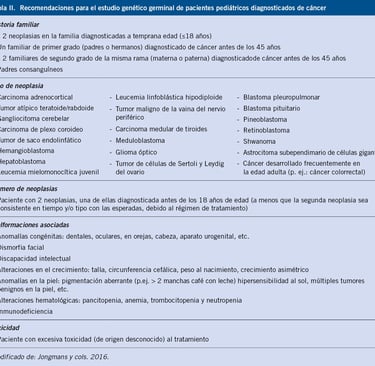
Tabla: pediatriaintegral.es
Síndromes de Predisposición al Cáncer Infantil
Algunos síndromes de predisposición al cáncer presentan características fenotípicas específicas que permiten a los médicos hacer un diagnóstico clínico temprano. Ejemplos de estos síndromes incluyen:
· Síndrome de Noonan: Asociado con una variedad de malformaciones congénitas, incluyendo problemas cardíacos, de crecimiento y faciales. Este síndrome aumenta el riesgo de leucemia.
· Neurofibromatosis tipo 1 (NF1): Este síndrome se asocia con un mayor riesgo de desarrollar neurofibromas, tumores en los nervios, y otros tipos de cáncer como gliomas.
· Síndrome de Beckwith-Wiedemann: Caracterizado por un crecimiento anormal y un mayor riesgo de desarrollar tumores abdominales, como el rabdomiosarcoma.
Los niños con tumores bilaterales, multifocales o metacrónicos también deben ser evaluados para detectar posibles síndromes de predisposición. Estos tumores pueden ser un indicador de trastornos subyacentes como los mencionados anteriormente.
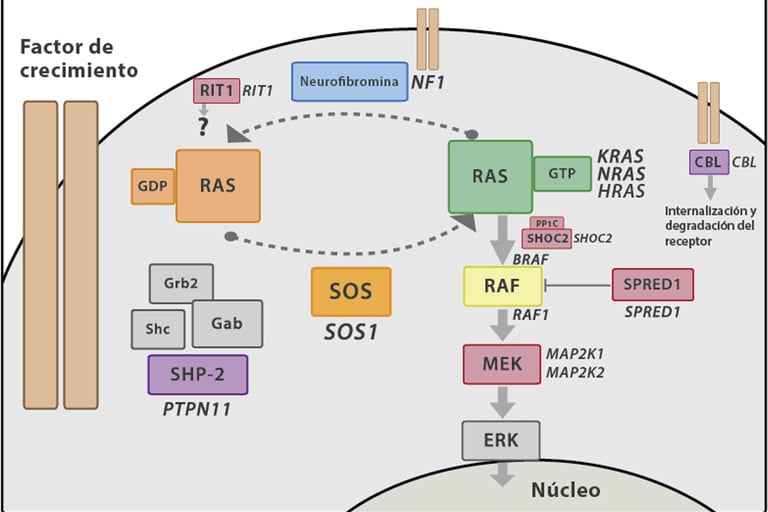
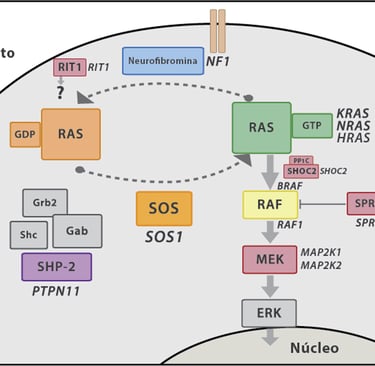
Imagen: Carcavilla et al. Síndrome de Noonan: actualización genética, clínica y de opciones terapéuticas. 2020. 93(1): 61.e1-61.e14
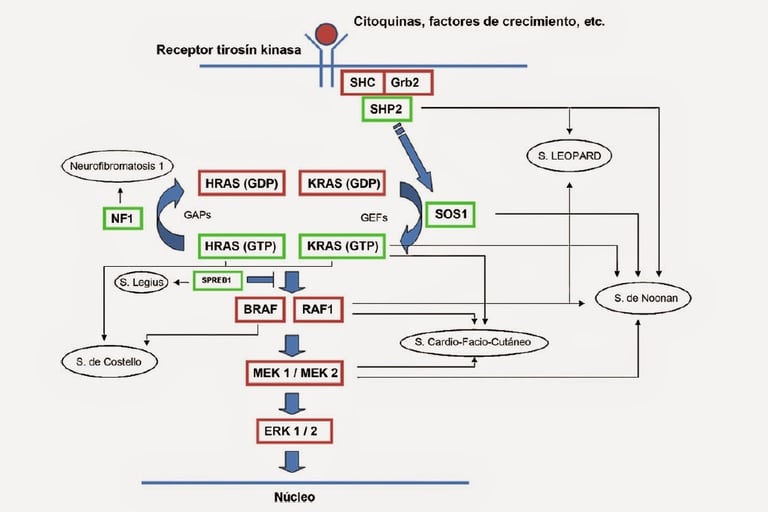
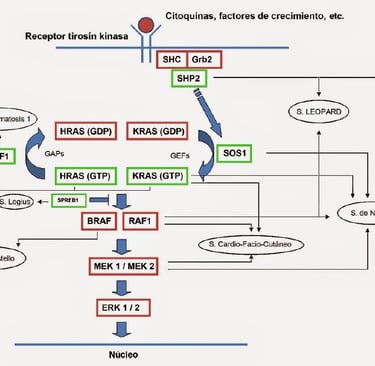
Imagen: dermapixel.com
Impacto de los Síndromes Genéticos en el Tratamiento
Es crucial reconocer que los niños con síndromes genéticos específicos, como la anemia de Fanconi o la ataxia telangiectasia, son más susceptibles a sufrir efectos adversos graves durante tratamientos como la quimioterapia o la radioterapia. Estos síndromes pueden afectar la capacidad del cuerpo para reparar el daño causado por estos tratamientos, lo que incrementa el riesgo de complicaciones graves y efectos a largo plazo.
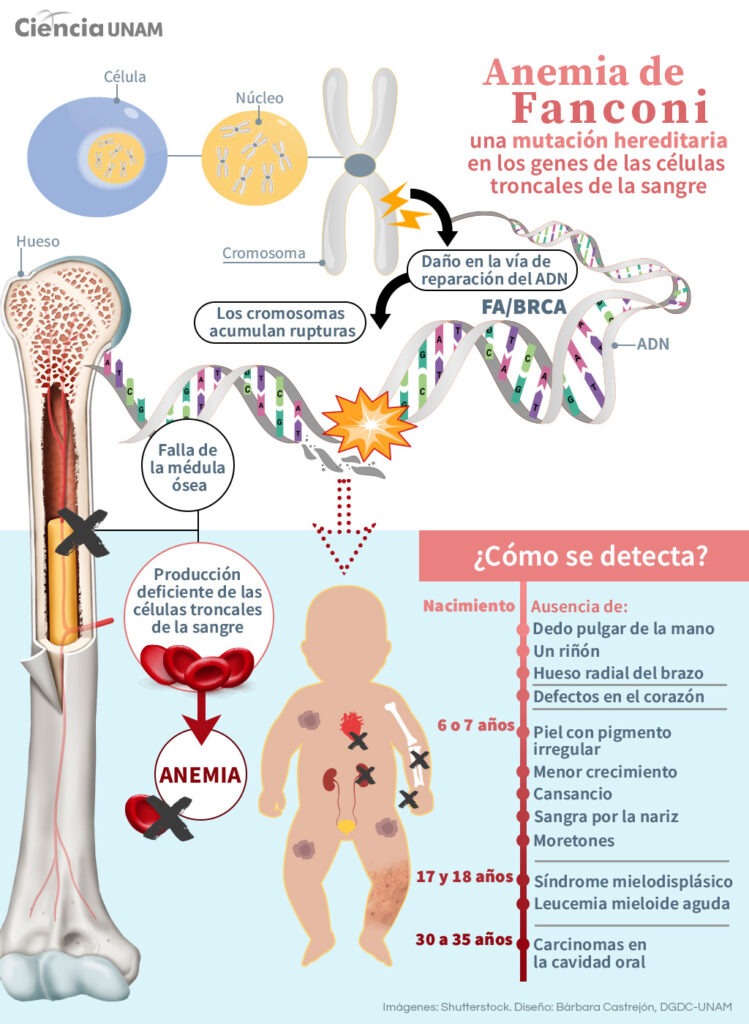
Imagen: unamglobal.unam.mx
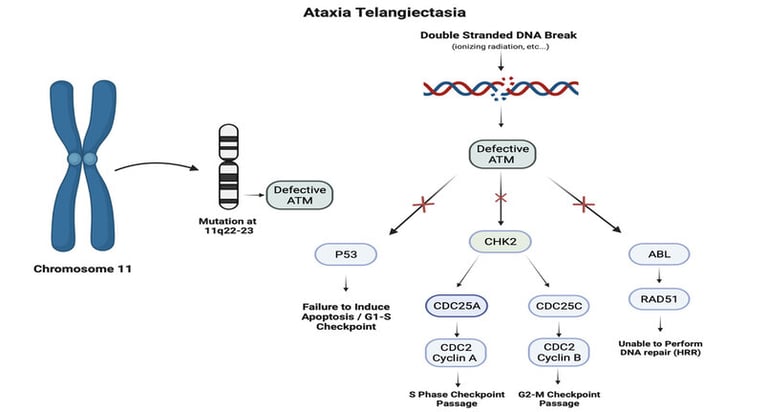
Imagen: Foster et al. Neurocutaneous diseases: Diagnosis, management and treatment. 2024
La Secuenciación de Nueva Generación (NGS) y su Relevancia Diagnóstica
El uso de la secuenciación de nueva generación (NGS) ha revolucionado el diagnóstico de predisposición genética al cáncer. Gracias a esta tecnología, los estudios han demostrado que hasta un 10% de los niños con cáncer presentan variantes patogénicas heredadas en genes de predisposición al cáncer. A través de NGS, los médicos pueden identificar mutaciones en genes clave, incluso antes de que se desarrollen características clínicas evidentes. Por ejemplo, el gen TP53, ubicado en el cromosoma 17 (locus 17p13.1), está relacionado con el síndrome de Li-Fraumeni, que se asocia con un alto riesgo de desarrollar varios tipos de cáncer a una edad temprana.
Este síndrome de herencia autosómica dominante está vinculado a un mayor riesgo de desarrollar una amplia gama de cánceres, como el carcinoma de corteza suprarrenal infantil, retinoblastoma, rabdomiosarcoma infantil, meduloblastoma y osteosarcoma pediátrico. Se estima que más del 1% de todos los casos de cáncer infantil están relacionados con variantes patogénicas de este gen.
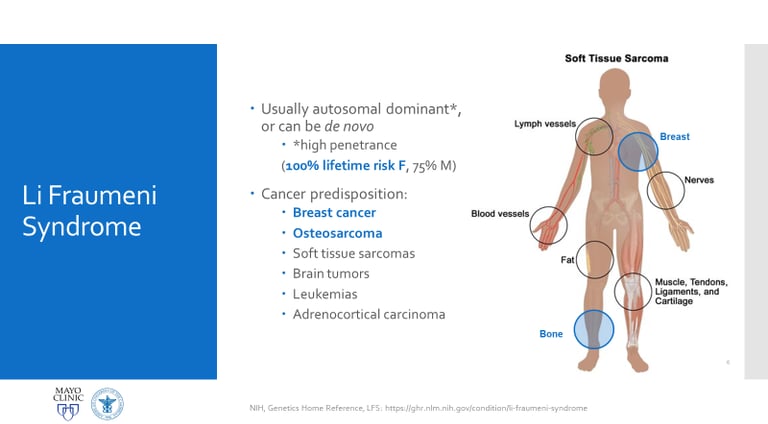
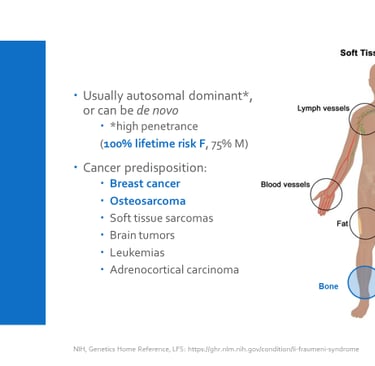
Imagen: labmedicineblog.com
El Papel de la Análisis Tumoral y la Interpretación Genética
Además de la secuenciación genética, el análisis de los tejidos tumorales es una herramienta crucial para el diagnóstico de síndromes de predisposición al cáncer. Por ejemplo, la pérdida de heterocigosidad en los genes supresores de tumores, como el gen BRCA1 en el cromosoma 17 (locus 17q21.31), puede ser un indicador clave de predisposición genética. También se observan marcas genéticas específicas, como la BRCAness, que sugiere una disfunción en la reparación del ADN.
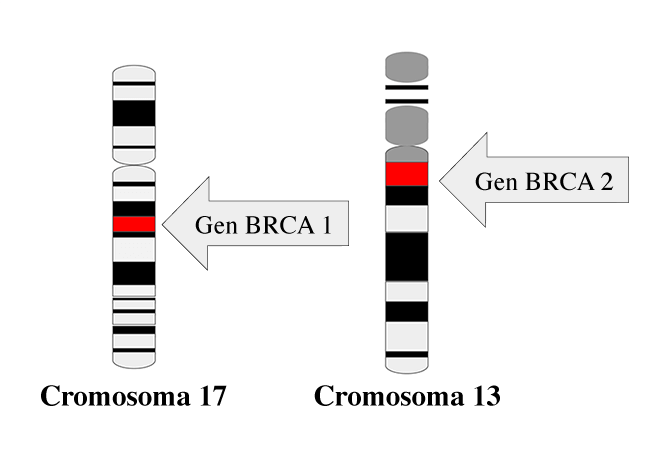
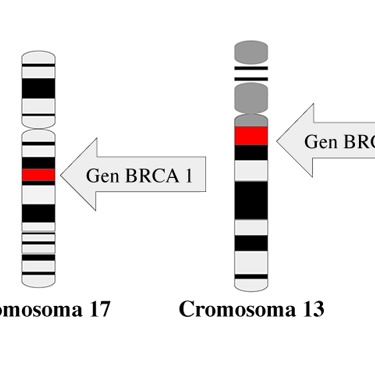
Imagen: national cancer institute
Desafíos y Futuro del Diagnóstico de Predisposición Genética
La detección de variantes de significación desconocida está aumentando debido al diagnóstico multimarcador. Esto requiere un enfoque diagnóstico integral que combine historia clínica detallada, análisis genético y exámenes laboratoriales exhaustivos, incluso cuando los pacientes no presentan características clínicas obvias. La colaboración entre centros especializados y la contribución a bases de datos públicas son esenciales para interpretar adecuadamente los resultados genéticos ambiguos y mejorar el diagnóstico de estos síndromes complejos.
Este enfoque integral permitirá avanzar en la identificación precoz de niños en riesgo de desarrollar cáncer, mejorando la prevención y el tratamiento personalizado, y proporcionando un pronóstico más preciso para aquellos que luchan contra el cáncer infantil.
Existen una gran variedad de tipos de cáncer infantil. A continuación se describirán una representación de los mismos:
Neoplasias hematológicas
Un aumento de condicionantes monogénicas se ha asociado con neoplasias mieloides y linfoblásticas. Algunas condiciones predisponentes han sido identificadas debido a su asociación con trastornos del desarrollo. Por ejemplo, el síndrome de Down está significativamente relacionado con un incremento de 500 veces con el riesgo de leucemia megacarioblástica aguda, que a menudo precede a una enfermedad mieloprolíferativa neonatal transitoria. Se ha visto también que un subconjunto de pacientes con el síndrome de Noonan desarrolló trastornos mieloprolifrativos neonatales, los cuales mejoran en la mayoría de los casos, aunque pueden ser mortales en otros.
Además, ambos síndromes aumentan el riesgo de leucemia linfoblástica aguda. Variantes en genes relacionados con la reparación del DNA, como el gen ATM y NBN, predisponen a las personas al desarrollo de cánceres linfoides, como la leucemia linfoblástica aguda de células T y el linfoma no Hodgkin, entre otros tipos de cáncer. El desarrollo de inmunodeficiencias es un rasgo distintivo de la mayoría de estos trastornos en la reparación del DNA.
Diversos estudios (Diaz-Flores et al., y Holmfeldt et al.) han revelado que el síndrome de Li Fraumeni se manifiesta en el 40% de los niños con leucemia linfoblástica hipodiploide aguda. Además, se han descubierto variantes hereditarias que afectan a factores de transcripción clave en las etapas reguladoras de la hematopoyesis. Estas variantes predisponen a los portadores a desarrollar neoplasias familiares (GATA2, RUNX1, ETV6, CEBPA) o leucemia linfoblástica aguda (RUNX1, ETV6, IKZF1, PAX5). Dichas variantes hereditarias pueden hacer que los afectados sean más sensibles a cambios a nivel somático, facilitando el desarrollo de subtipos específicos de leucemia, como es el caso de las asociadas con ETV6 y leucemia linfoblástica hiperdiploide.
En resumen, los síndromes de predisposición a neoplasias hematológicas representan una combinación única de factores genéticos y ambientales que requieren atención multidisciplinar para maximizar el beneficio para los pacientes.
Tumor cerebral
Se calcula que aproximadamente el 8% de los tumores cerebrales en niños se deben a mutaciones hereditarias en genes asociados con el cáncer. Algunos tipos específicos de tumores cerebrales están relacionados con un porcentaje más alto de síndromes de predisposición al cáncer, que suelen tener una alta penetrancia. Por ejemplo, los tumores rabdoides (SMARCB1) y los carcinomas del plexo coroideano (TP53) se han relacionado con un elevado porcentaje de predisposición al cáncer.
Se han detectado altos índices de mutaciones en genes de la línea germinal relacionados con el cáncer, como son NF2, LZTR1, SMARCB1 o SMARCE1, en casos de meningiomas y schwannomas que aparecen antes de los 25 años. En pacientes con meduloblastoma, aproximadamente el 6% porta una mutación hereditaria en un gen relacionado con el cáncer. En algunos subgrupos específicos de meduloblastoma, esta tasa es significativamente más alta, lo que refleja un mayor riesgo genético de desarrollar cáncer en estas poblaciones.
En niños con meduloblastoma donde la vía de regulación Sonic hedgehog está afectada, los diagnosticados a una edad mayor (de 5 a 15 años) tienen una alta probabilidad de portar una mutación hereditaria en TP53, mientras que los diagnosticados a una edad más joven (0-3 años) suelen presentar una mutación hereditaria en el gen SUFU (síndrome de Gorlin). Posteriormente, se identificaron_mutaciones de pérdida de función en el gen ELP1 en el 14% de pacientes con meduloblastoma con la vía de regulación Sonic hedgehog afecto; ELP1 codifica una subunidad del complejo elongador relacionada con la homeostasis proteica. Además, se identificaron variantes en GPR161 en casos raros de este tipo de meduloblastoma
Los niños con meduloblastoma inducido por WNT que no presentan variants somáticas en CTNNB1 generalmente portan mutaciones hereditarias en el gen APC.
El glioma de alto grado infantil está relacionado con el síndrome de Li Fraumeni y la enfermedad de defecto hereditario en el proceso de reparación por mismatch. En esta enfermedad, se observan tipos inmunohistoquímicos específicos, perfiles inmunológicos específicos y un fenotipo hipermutaador que sugieren el síndrome de Li Fraumeni. Además, los gliomas cerebrales extremadamente hipermutadores han sido asociados con variantes patógenas en la polimerasa de la reparación por mismatch (POLE). En dos casos analizados, uno con meduloblastoma y otro con glioma de alto grado hipermutado, se han identificado estos vínculos genéticos. Por otro lado, el glioma de bajo grado se asocia con neurofibromatosis tipo 1 y a veces con otras patologías relacionadas con la vía de activación RAS.
Blastomas
Aproximadamente un 10% de los casos de retinoblastoma se produce en pacientes con predisposición en la línea germinal, aunque en la mayoría de las veces no se identifican mutationes hereditarias específicas. Las probabilidades aumentan significativamente si hay un historial familiar de cáncer de mama o de ovario, así como de otros tipos de cáncer relacionados con el desarrollo genital.
El tumor de Wilms, también conocido como nefroblastoma, se considera un ejemplo primordial de cómo los procesos anómalos pueden relacionarse con la carcinogénesis. El 10-15% de los niños que padecen este tumor tienen un síndrome de predisposición genética al cáncer, aunque esta variación puede estar relacionado con el origen del paciente.
Entre los genes involucrados se encuentran WT1, TRIM28, REST y CTR9, que son específicos del tumor de Wilms, así como otros genes como PALB2 o BLM. Los genes como TRIM28 y REST rara vez mutan somáticamente, lo que indica que sus efectos oncogénicos se restringen a etapas de desarrollo tempranas. El diagnóstico del tumor de Wilms bilateral muestra una distribución bimodal de la edad, lo que sugiere una complejidad genética que no se comprende plenamente. No todos los pacientes portan genes conocidos que predispongan su desarrollo, lo que indica la existencia de factores genéticos adicionales. En un 12% de los casos bilaterales, se han observado cambios epigenéticos y mutaciones heredables que afectan a H19 o IGF2. Aunque algunos casos pueden explicarse debido a un mosaicismo somático temprano, todos los pacientes con tumor bilateral de Wilms deben ser investigados como potenciales portadores de genes que predispongan al cáncer, los cuales podrían transmitirse a la descendencia.
La presunción de predisposición se ve incrementada por la edad joven, la presentación bilateral o multifocal del tumor, anomalías congénitas (especialmente urológicas), síndromes de crecimiento excesivo y tipos histológicos específicos como el subtipo estromal en mutantes de WT1 y el tumor epitelial diferenciado en mutantes de TRIM28. Aunque las tasas de supervivencia son altas, es recomendable realizar consejo genético y estudios para evaluar los riesgos, especialmente en hermanos y futuras generaciones.
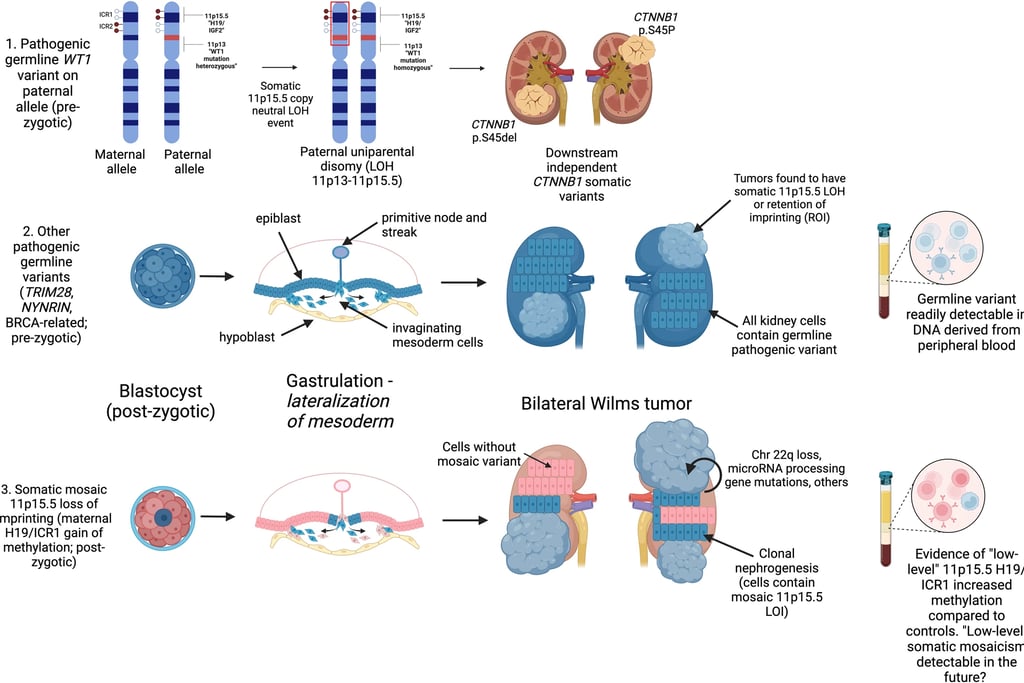
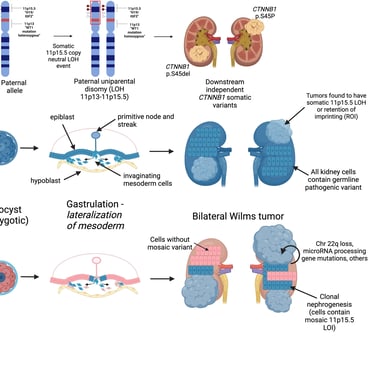
Imagen: Murphy et al. Genetic and epigenetic features of bilateral Wilms tumor predisposition in patients from the Children’s Oncology Group AREN18B5-Q. Nat Commun 14, 8006 (2023)
El hepatoblastoma es un tumor hepático raro en la infancia, representando aproximadamente el 1% de los cánceres pediátricos. Su desarrollo está asociado con condiciones genéticas como el síndrome de Beckwith-Wiedemann y la poliposis adenomatosa familiar, vinculada a mutaciones en el gen APC (ubicado en el locus 5q22.2). Aunque el síndrome de Beckwith-Wiedemann suele diagnosticarse por sus rasgos clínicos, existen casos en los que no se observan características fenotípicas evidentes, pero sí alteraciones epigenéticas en el locus 11p15.5, detectables mediante estudios moleculares.
En niños con hepatoblastoma que no presentan mutaciones somáticas en el gen CTNNB1 (locus 3p22.1), se recomienda realizar pruebas genéticas para detectar mutaciones en APC, ya que estas pueden aparecer de manera espontánea sin antecedentes familiares de poliposis. Dado que las mutaciones en APC y CTNNB1 son excluyentes entre sí, el análisis del gen APC puede limitarse a aquellos casos en los que no se identifican alteraciones en CTNNB1.
Los análisis completos del exoma de ADN de grandes grupos de pacientes sugieren que los neuroblastomas esporádicos, en su mayoría, no están relacionados con síndromes genéticos que aumenten el riesgo de cáncer, incluso en personas jóvenes. Es importante investigar mutaciones en los genes ALK (locus 2p23.1) y PHOX2B (locus 3p11.1), que son los principales responsables de la predisposición al neuroblastoma, especialmente en casos familiares o cuando el cáncer afecta a varias áreas. Las asociaciones con síndromes genéticos son poco comunes. También se ha encontrado una relación entre el riesgo de neuroblastoma y ciertas variantes en el gen TP53 (locus 17p13.1), así como con polimorfismos genéticos que afectan la susceptibilidad. Algunos de estos polimorfismos modifican regiones del ADN que actúan como reguladores en el gen LMO1 (locus 11p15.5), el cual es clave para controlar la sobreexpresión del gen MYCN (locus 2p24.3). Este tipo de variantes, que se encuentran en áreas no codificantes del ADN, muestra cómo las interacciones entre eventos genéticos adquiridos y las regiones no codificantes del genoma pueden influir en la susceptibilidad a neuroblastomas de alto riesgo.
Sarcoma
Varios síndromes de predisposición genética están asociados con un mayor riesgo de sarcomas específicos y tumores de tejidos blandos. Probablemente, la asociación más conocida de un síndrome de predisposición al cáncer con sarcoma es el síndrome de Li-Fraumeni. El osteosarcoma es la malignidad más frecuente en niños con síndrome de Li-Fraumeni (30% de todos los tumores), seguido de los sarcomas de tejidos blandos (23% de todos los tumores). Otro caso conocido, aunque raro, de osteosarcoma son las variantes bialélicas en las helicasas de ADN, como BLM (locus 15q26.1), que causa el síndrome de Bloom, y RECQL4 (locus 8q24.3), que causa el síndrome de Rothmund-Thomson.
Se ha demostrado que las variantes heterocigotas en el gen RECQL4 (locus 8q24.3) están significativamente sobre representadas en niños con osteosarcoma. El osteosarcoma es un caso raro de cáncer para el cual se puede predecir el riesgo de desarrollarlo en la infancia mediante determinación poligénica. A través del análisis de más de 400 polimorfismos de nucleótido simple asociados con la altura alcanzada en la infancia y la adultez, Zhang et al. desarrollaron puntuaciones poligénicas que se relacionaron tanto con una mayor estatura como con un aumento en el riesgo de osteosarcoma. Este ejemplo ilustra cómo los estudios de asociación a nivel genómico arrojan nueva luz sobre observaciones clínicas antiguas, como la posible relación entre una mayor estatura y el osteosarcoma.
Se ha demostrado que las mutaciones germinales heterocigotas patogénicas (o potencialmente patogénicas) en varios genes de reparación del ADN están enriquecidas en una cohorte de pacientes con sarcoma de Ewing. Los autores sugieren que estas mutaciones germinales heterocigotas podrían provocar rupturas en el ADN y, posteriormente, la fusión entre el gen EWSR1 (locus 22q12.2) y los genes que codifican la familia de factores de transcripción ETS (fusión EWSR1–ETS), una hipótesis que podría ser relevante para otros tipos de cáncer y que debe ser investigada más a fondo. Los estudios de asociación a nivel genómico también han resaltado la cooperación entre los microsatélites polimórficos constitucionales y las fusiones adquiridas EWSR1–ETS; el reemplazo de una A por una T en la región potenciadora del gen EGR2 (locus 6q23.3) afecta sustancialmente la capacidad de las proteínas quiméricas EWSR1–ETS para unirse a esta región reguladora y, por lo tanto, ejercer sus efectos oncogénicos. Este ejemplo ilustra nuevamente el papel de la parte no codificante del genoma en la susceptibilidad al cáncer infantil y puede explicar en parte por qué el sarcoma de Ewing es más prominente en pacientes de ascendencia europea blanca que en otros orígenes étnicos.
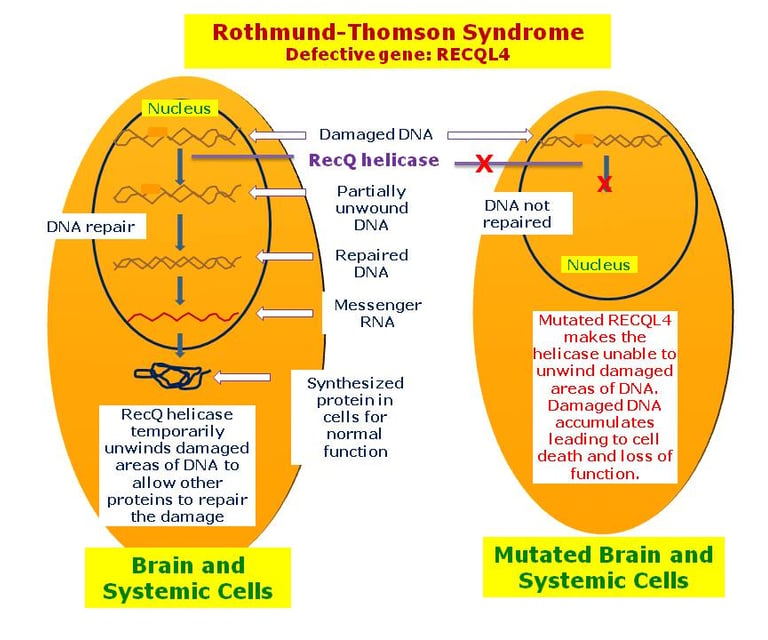
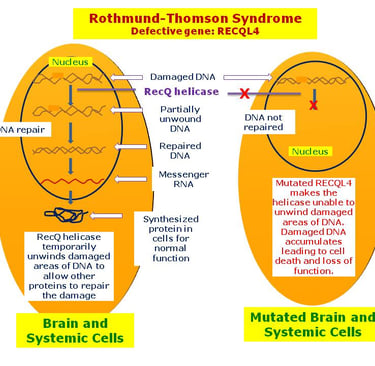
imagen: disorders.eyes.arizona.edu
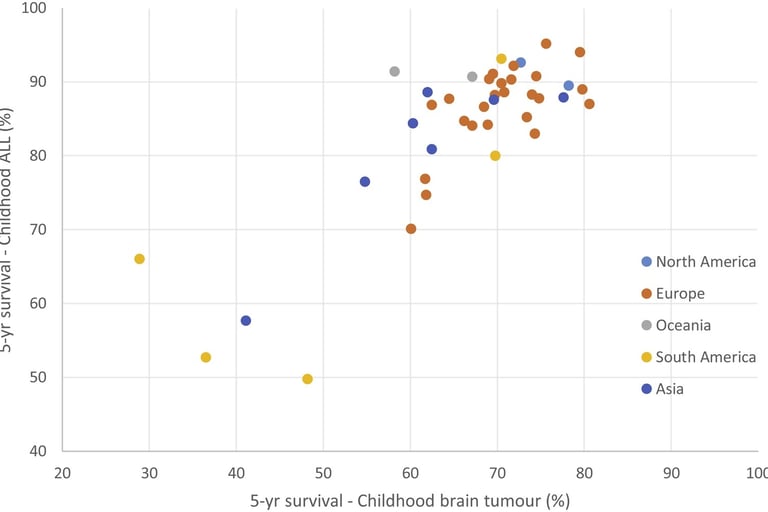
imagen: Erdmann et al. Childhood cancer: Survival, treatment modalities, late effects and improvements over time. Cancer Epidemiology. 2021
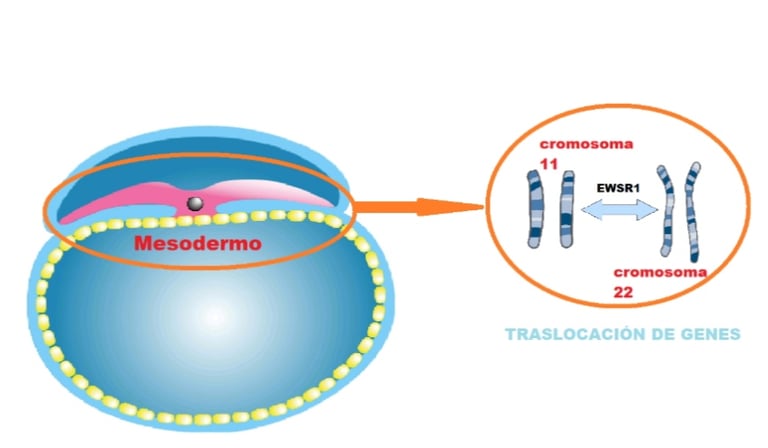
imagen: microbacterium.es
Tratamientos en Cáncer Infantil: Avances y Desafíos en la Medicina de Precisión
Recientes avances en la medicina de precisión han transformado el panorama del tratamiento del cáncer infantil, especialmente en los casos de cánceres de alto riesgo. La incorporación de herramientas usualmente usadas en el diagnóstico, como la secuenciación de próxima generación (NGS), y el desarrollo de terapias dirigidas han permitido identificar nuevas estrategias de tratamiento. Estos avances han mostrado un potencial considerable en la personalización de los tratamientos, basados en las características moleculares de cada tumor. Sin embargo, a pesar de los progresos, la adopción de terapias dirigidas sigue siendo limitada, con tasas de aceptación que oscilan entre el 10% y el 33%. Esto se debe principalmente a la incertidumbre sobre la efectividad de estas terapias y a la dificultad para equilibrar los beneficios y riesgos asociados.
Medicina de Precisión y Terapias dirigidas
En oncología pediátrica, los avances en la medicina de precisión han permitido identificar objetivos moleculares en más del 65% de los niños con cánceres de alto riesgo. Los estudios como INFORM y GAIN han demostrado que las respuestas de los tumores pueden estar relacionadas con fusiones genéticas activadoras específicas. Por ejemplo, ciertas fusiones de genes, como ALK o ROS1, se asocian con una mejor respuesta a terapias dirigidas.
A pesar de estos avances, aún se desconoce cuáles pacientes se beneficiarán más de las terapias guiadas por precisión. La secuenciación de próxima generación ha permitido identificar mutaciones en genes claves, lo que abre la puerta a tratamientos más específicos. En estudios como PRISM (ZERO Childhood Cancer Precision Medicine Program), se identificaron objetivos moleculares en los tumores de los pacientes y se ofrecieron tratamientos personalizados, con un seguimiento de al menos 18 meses.
Terapias guiadas por precisión (PGT) vs no guiadas o tratamientos estándar: comparación de supervivencia y respuesta al tratamiento
En un análisis comparativo entre pacientes tratados con terapias guiadas por precisión (PGT) y aquellos tratados con terapias no dirigidas (como tratamientos estándar), los resultados de supervivencia libre de progresión (PFS) fueron significativamente mejores en los primeros. Por ejemplo, los pacientes tratados con PGT mostraron una PFS a dos años de un 27%, en comparación con el 11% en aquellos que recibieron tratamientos no guiados.
No obstante, la diferencia en la supervivencia global (OS) no alcanzó significancia estadística en todos los casos, lo que plantea la necesidad de más estudios para evaluar el impacto a largo plazo de estas terapias personalizadas.
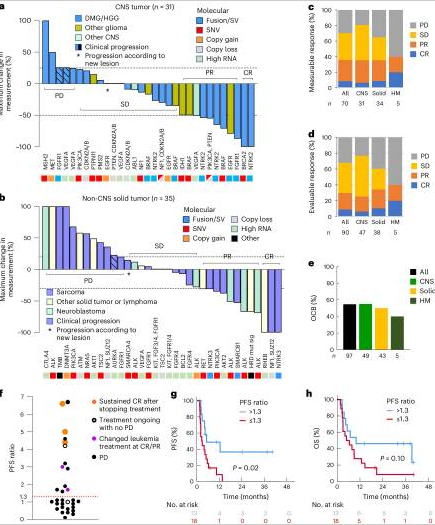
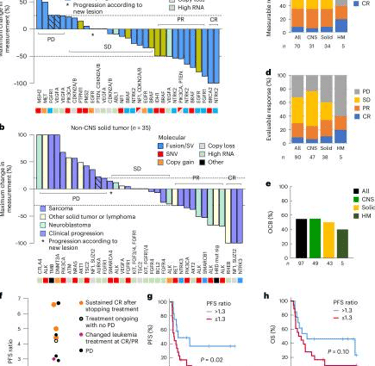
Figura: Lau LMS et al. Precision-guided treatment in high-risk pediatric cancers. Nat Med. 2024 Jul;30(7):1913-1922.
Respuesta a Tratamientos Dirigidos: Fusiones y Mutaciones Genéticas
El éxito de las terapias dirigidas varió según las alteraciones genéticas presentes en los tumores de los pacientes. Las fusiones génicas y las variaciones estructurales (SV) fueron las que mostraron una tasa de respuesta más alta, con un 60% de los pacientes respondiendo favorablemente a tratamientos dirigidos. En comparación, las mutaciones de un solo nucleótido (SNV) tuvieron una tasa de respuesta de un 32%, mientras que otras alteraciones, como las variaciones en el número de copias de genes (CNV), tuvieron tasas de respuesta de solo un 14%.
Por ejemplo, fusión ALK y ROS1 fueron objetivos clave en pacientes con cáncer de pulmón pediátrico y neuroblastoma, mostrando una tasa de respuesta notablemente alta cuando se dirigieron con tratamientos específicos.
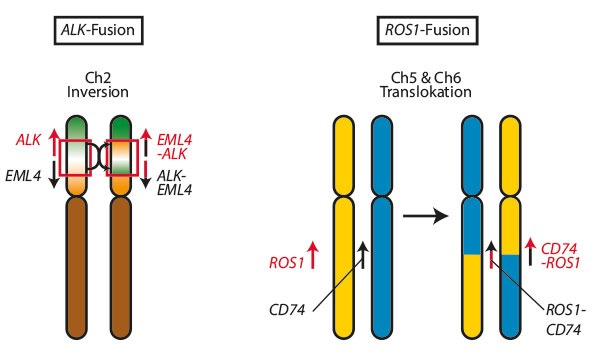
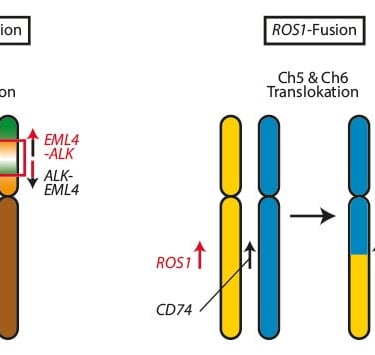
Imagen: Trillium.de
Desafíos y Futuro de las Terapias Dirigidas: Limitaciones en el acceso a medicamentos y ensayos clínicos
A pesar de los prometedores resultados de la medicina de precisión, existen desafíos importantes en su implementación generalizada. El acceso a fármacos dirigidos específicos para alteraciones genómicas pediátricas y la disponibilidad de ensayos clínicos adecuados sigue siendo limitado, lo que obstaculiza el beneficio para muchos pacientes. Se necesita una mayor inversión en el desarrollo de terapias dirigidas específicas para los niños y en el diseño de ensayos clínicos más inclusivos.
En resumen, la medicina de precisión ha mejorado significativamente las opciones de tratamiento para los niños con cánceres de alto riesgo, especialmente aquellos con alteraciones genéticas bien establecidas. Sin embargo, los desafíos en la implementación de estas terapias y la falta de datos definitivos sobre su impacto en la supervivencia global siguen siendo puntos críticos. A medida que más estudios se centren en las fusiones genéticas, las mutaciones de un solo nucleótido y otros cambios moleculares específicos, es probable que veamos un avance continuo en la eficacia de estos tratamientos. La integración de la medicina personalizada en la oncología pediátrica es fundamental para mejorar la supervivencia de los niños con cáncer.
Referencias:
Erdmann, F. et al. Childhood cancer: Survival, treatment modalities, late effects and improvements over time. 2021. Cancer Epidemiology. 71b: 101733
Lau LMS et al.Precision-guided treatment in high-risk pediatric cancers. Nat Med. 2024 Jul;30(7):1913-1922
Kratz, Christian P et al.Predisposition to cancer in children and adolescents. The Lancet Child & Adolescent Health, Volume 5, Issue 2, 142 - 154
Nakano Y, Rabinowicz R, Malkin D. Genetic predisposition to cancers in children and adolescents. Curr Opin Pediatr. 2023 Feb 1;35(1):55-62.
Roganovic J. Genetic predisposition to childhood cancer. World J Clin Pediatr. 2024 Sep 9;13(3):95010
PDQ Cancer Genetics Editorial Board. Cancer Genetics Risk Assessment and Counseling (PDQ®): Health Professional Version. 2025 Jan 3.
Orbach D., Brech B. I. et al. The role of cancer predisposition syndrome in children and adolescents with very rare tumours. 2023. EJC Paediatric Oncology. 2: 100023

(anuncios)