


Childhood Cancer: A Global Perspective and Its Challenges
José Hernández Jiménez
2/14/202515 min read


Childhood Cancer: A Global Perspective and Its Challenges
Introduction
Childhood cancer is a diverse group of malignant neoplasms that encompasses a variety of diseases with very different characteristics in terms of onset, causes, treatment, care, and prognosis. Despite being a heterogeneous group, research and medical advancements have significantly improved survival rates in recent decades, transforming the outlook for many children affected by this disease.
Advances in Treatment and Survival
Globally, advances in childhood cancer research have led to significant improvements in treatments and survival rates. In developed countries, such as the United States, 85% of children diagnosed with cancer survive for more than five years. However, in other parts of the world, especially in regions of Africa, Latin America, and Asia, survival rates are considerably lower. For example, in some areas of East Africa, these rates may be as low as 8%. These disparities in survival reflect differences in access to specialized treatments, medical resources, and the training of healthcare professionals.
Geographical Inequality in Survival Rates
Survival rates vary not only between countries but also within the more developed regions, where not all types of childhood cancer present the same prognosis. Acute lymphoblastic leukemia (ALL) has better outcomes compared to other types, such as acute myeloid leukemia (AML). Additionally, diseases like Burkitt lymphoma or Hodgkin lymphoma have even higher survival rates. However, the differences in survival are not only due to the type of cancer but also to social and geographical factors, highlighting the persistent inequality in access to healthcare.
Socioeconomic and Demographic Factors in Prognosis
Age, sex, and socioeconomic status play a crucial role in the outcomes of childhood cancer patients. Neonates and infants tend to have better survival prospects in certain types of cancer. However, the socioeconomic status of families continues to negatively influence outcomes, even in countries with advanced healthcare systems. This highlights the importance of addressing both social and economic factors to ensure equitable care for all children affected by this disease.
Side Effects and Quality of Life of Survivors
Childhood cancer treatment not only focuses on eradicating the disease but also on the long-term side effects that children may experience. Persistent pain during and after treatment is one of the most common issues, affecting the quality of life of survivors. Additionally, children who survive cancer may face premature aging and increased physical fragility, which raises the risk of chronic diseases, falls, and hospitalizations in the future. This accelerated aging is not exclusive to childhood cancer survivors but also affects others with chronic diseases related to aging.
Challenges and Future Outlook
Despite advancements in treatment and improved survival rates, childhood cancer still presents significant challenges globally. Inequalities in access to healthcare and the side effects of treatment are issues that still require attention. Ongoing research and international collaboration are essential to reduce these disparities, improve the quality of life of survivors, and ultimately make progress toward curing these diseases.
This article addresses the main challenges and advancements in childhood cancer treatment, highlighting the urgent need for equitable and sustainable solutions in access to healthcare for all children, regardless of their place of residence or economic situation.
Diagnosis and Genetic Predisposition to Childhood Cancer
The diagnosis of childhood cancer with genetic predisposition involves a series of tests and clinical evaluations focused on the family history and the patient's phenotypic characteristics. In addition, genetics plays a crucial role, as certain genetic variants can increase the risk of developing cancer at an early age. This section discusses diagnostic methods and the main childhood cancer predisposition syndromes, as well as the impact of genetics on treatment.
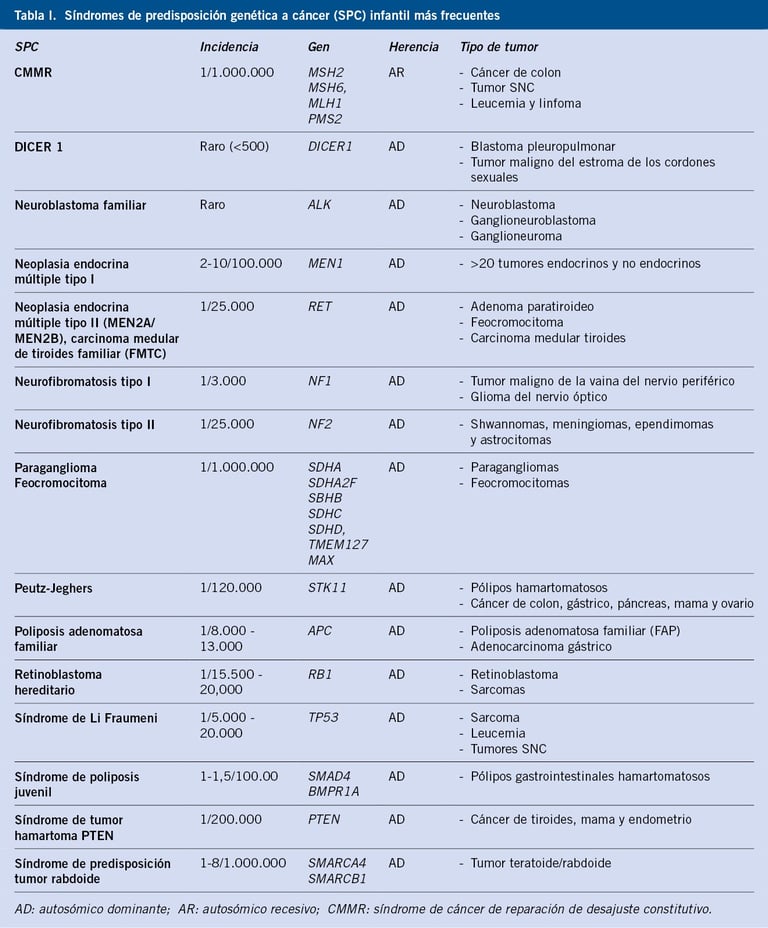
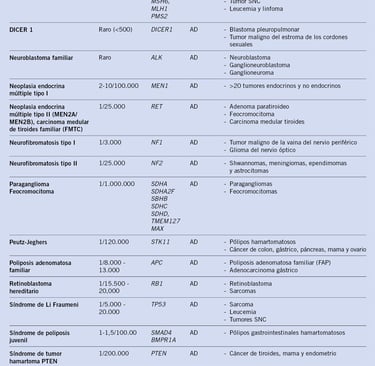
Table: pediatriaintegral.es
Clinical Diagnosis and Family History Assessment
Traditionally, the diagnosis of childhood cancer predisposition syndromes is based on clinical suspicion. Doctors typically begin by evaluating the family history, looking for patterns that suggest a hereditary predisposition. It is important to include information from at least three generations, as this may indicate an inherited genetic mutation. However, a negative family history does not necessarily exclude the diagnosis, as there are de novo mutations (new genetic variants not inherited from the parents) and other factors, such as reduced penetrance, that may not be evident in the family history.
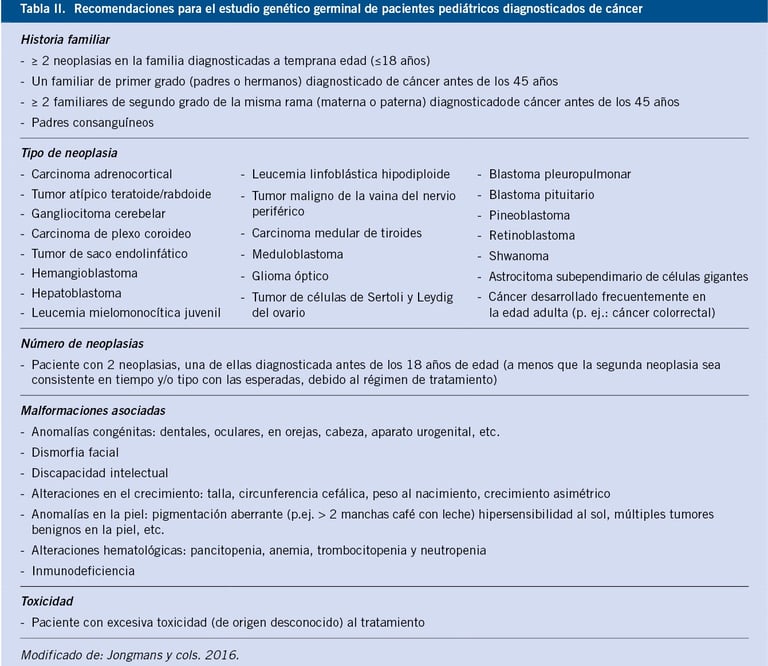
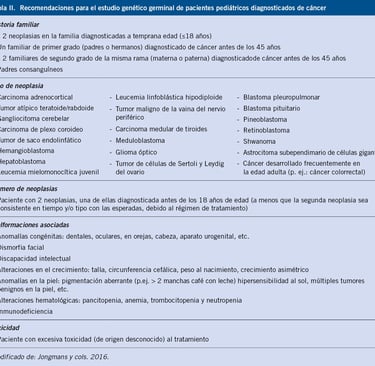
Table: pediatriaintegral.es
Childhood Cancer Predisposition Syndromes
Some cancer predisposition syndromes present specific phenotypic characteristics that allow doctors to make an early clinical diagnosis. Examples of these syndromes include:
Noonan Syndrome: Associated with a variety of congenital malformations, including heart, growth, and facial problems. This syndrome increases the risk of leukemia.
Neurofibromatosis Type 1 (NF1): This syndrome is associated with a higher risk of developing neurofibromas, tumors on the nerves, and other types of cancer such as gliomas.
Beckwith-Wiedemann Syndrome: Characterized by abnormal growth and an increased risk of developing abdominal tumors, such as rhabdomyosarcoma.
Children with bilateral, multifocal, or metachronous tumors should also be evaluated for possible predisposition syndromes. These tumors can be an indicator of underlying disorders, such as those mentioned above.
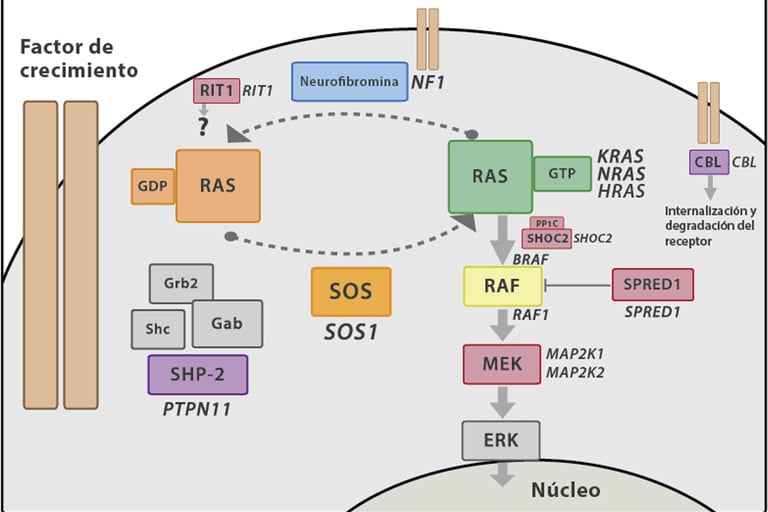
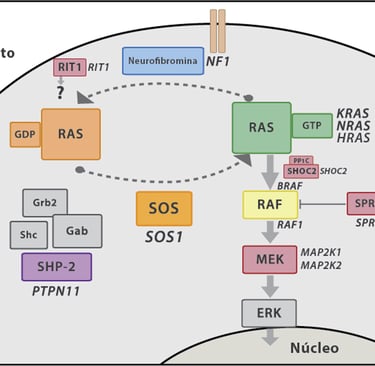
Imagen: Carcavilla et al. Síndrome de Noonan: actualización genética, clínica y de opciones terapéuticas. 2020. 93(1): 61.e1-61.e14
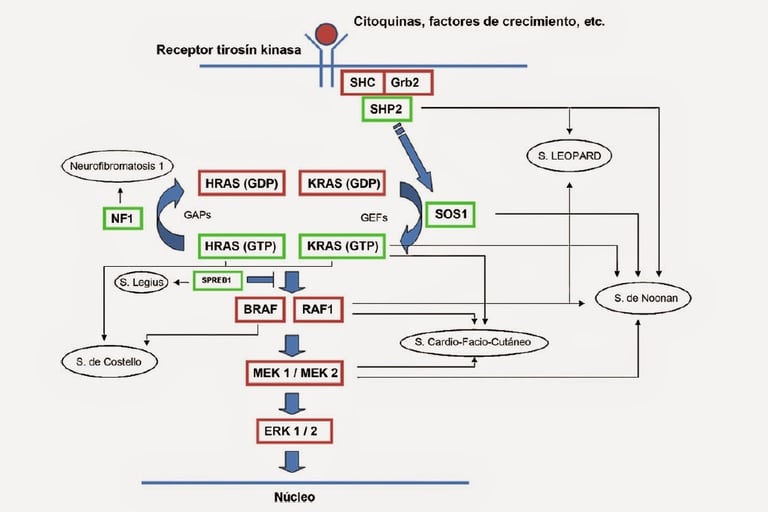
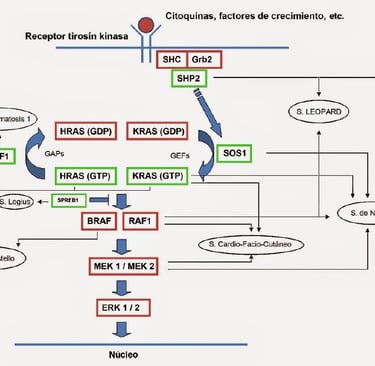
Imagen: dermapixel.com
Impact of Genetic Syndromes on Treatment
It is crucial to recognize that children with specific genetic syndromes, such as Fanconi anemia or ataxia telangiectasia, are more susceptible to experiencing severe adverse effects during treatments like chemotherapy or radiation therapy. These syndromes can affect the body's ability to repair the damage caused by these treatments, increasing the risk of serious complications and long-term effects.
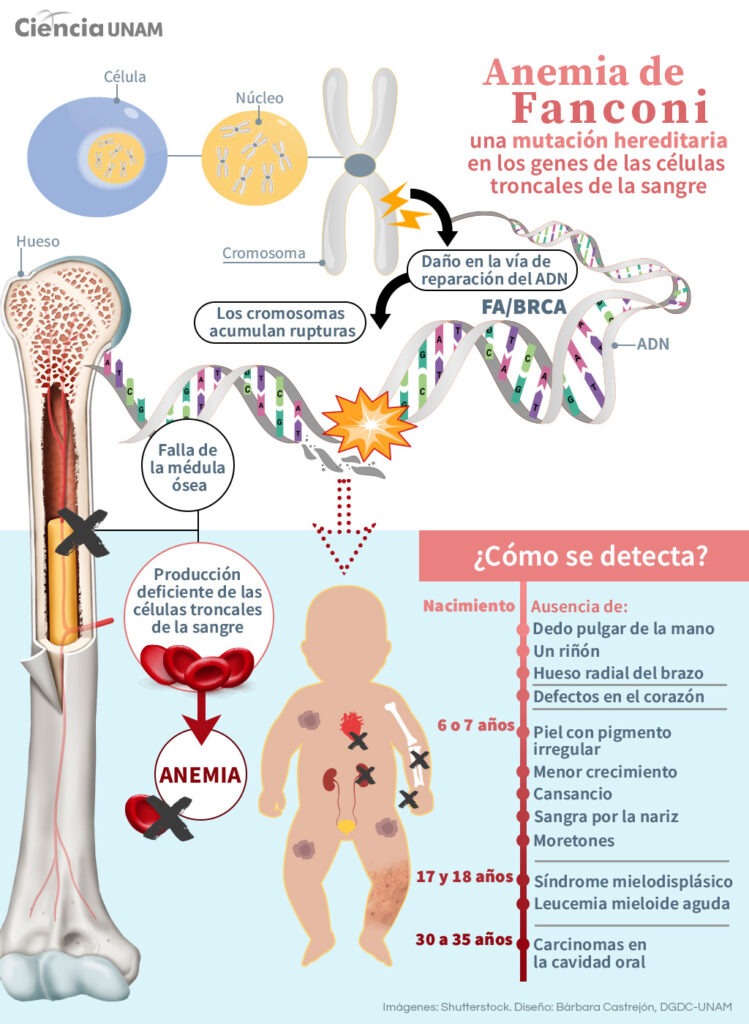
Image: unamglobal.unam.mx
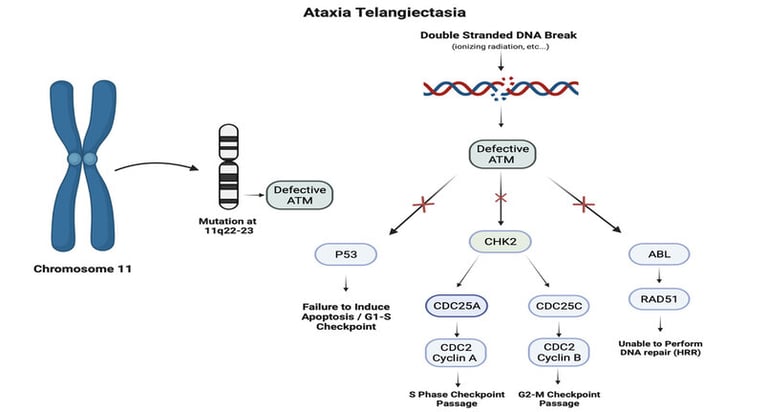
Image: Foster et al. Neurocutaneous diseases: Diagnosis, management and treatment. 2024
Next-Generation Sequencing (NGS) and Its Diagnostic Relevance
The use of next-generation sequencing (NGS) has revolutionized the diagnosis of genetic predisposition to cancer. Thanks to this technology, studies have shown that up to 10% of children with cancer have inherited pathogenic variants in cancer predisposition genes. Through NGS, doctors can identify mutations in key genes, even before obvious clinical characteristics develop. For example, the TP53 gene, located on chromosome 17 (locus 17p13.1), is associated with Li-Fraumeni syndrome, which is linked to a high risk of developing various types of cancer at an early age.
This autosomal dominant inherited syndrome is associated with a higher risk of developing a wide range of cancers, such as childhood adrenal cortex carcinoma, retinoblastoma, childhood rhabdomyosarcoma, medulloblastoma, and pediatric osteosarcoma. It is estimated that more than 1% of all childhood cancer cases are related to pathogenic variants of this gene.
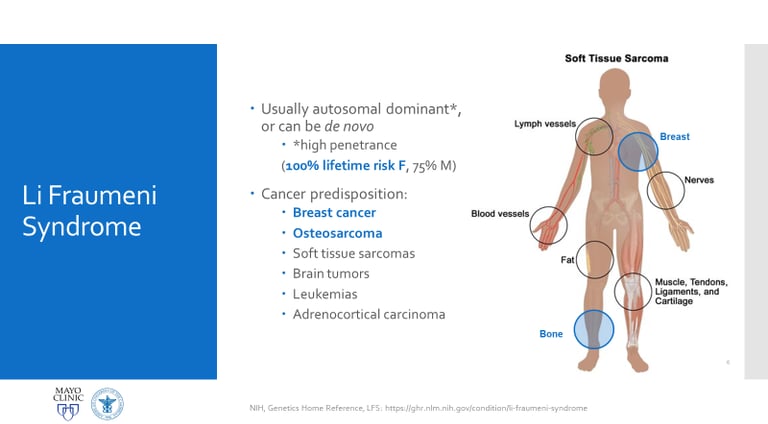
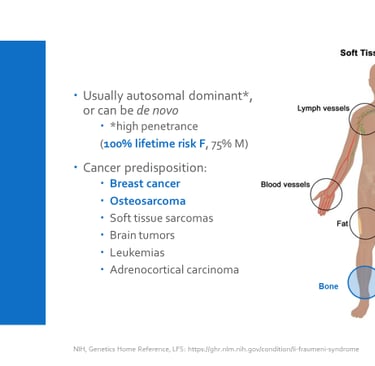
Image: labmedicineblog.com
The Role of Tumor Analysis and Genetic Interpretation
In addition to genetic sequencing, tumor tissue analysis is a crucial tool for diagnosing cancer predisposition syndromes. For example, loss of heterozygosity in tumor suppressor genes, such as the BRCA1 gene on chromosome 17 (locus 17q21.31), can be a key indicator of genetic predisposition. Specific genetic marks, such as BRCAness, are also observed, suggesting a dysfunction in DNA repair.
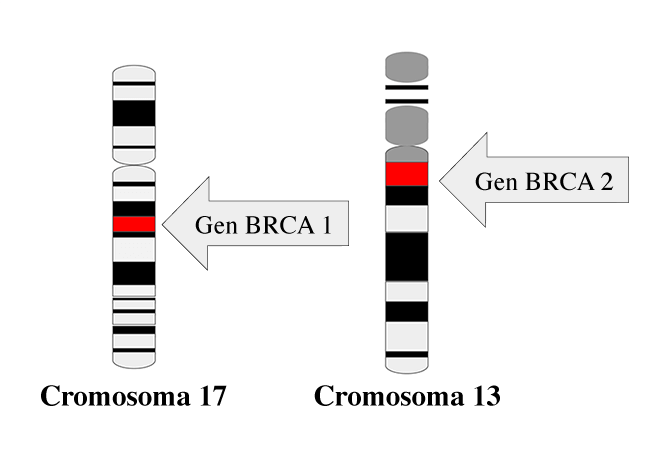
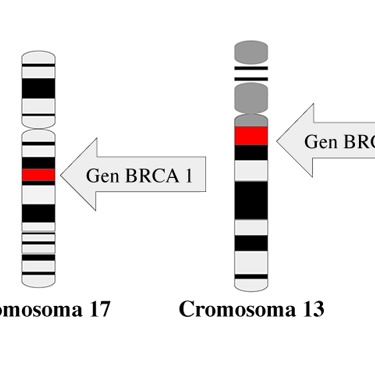
Image: national cancer institute
Challenges and Future of Genetic Predisposition Diagnosis
The detection of variants of unknown significance is increasing due to multimarker diagnosis. This requires a comprehensive diagnostic approach that combines detailed clinical history, genetic analysis, and thorough laboratory tests, even when patients do not present obvious clinical features. Collaboration between specialized centers and contributions to public databases are essential to properly interpret ambiguous genetic results and improve the diagnosis of these complex syndromes.
This integrated approach will enable progress in the early identification of children at risk of developing cancer, enhancing prevention and personalized treatment, and providing a more accurate prognosis for those fighting childhood cancer.
There is a wide variety of types of childhood cancer. Below is a representation of these types:
Hematological Neoplasms
An increase in monogenic conditions has been associated with myeloid and lymphoblastic neoplasms. Some predisposition conditions have been identified due to their association with developmental disorders. For example, Down syndrome is significantly associated with a 500-fold increased risk of acute megakaryoblastic leukemia, which often precedes transient neonatal myeloproliferative disease. It has also been observed that a subset of patients with Noonan syndrome developed neonatal myeloproliferative disorders, which improve in most cases, although they can be fatal in others.
Furthermore, both syndromes increase the risk of acute lymphoblastic leukemia. Variants in genes related to DNA repair, such as the ATM and NBN genes, predispose individuals to the development of lymphoid cancers, such as T-cell acute lymphoblastic leukemia and non-Hodgkin lymphoma, among other types of cancer. The development of immunodeficiencies is a distinctive feature of most of these disorders in DNA repair.
Various studies (Diaz-Flores et al., and Holmfeldt et al.) have revealed that Li-Fraumeni syndrome manifests in 40% of children with hypodiploid acute lymphoblastic leukemia. Additionally, hereditary variants affecting key transcription factors in the regulatory stages of hematopoiesis have been discovered. These variants predispose carriers to develop familial neoplasms (GATA2, RUNX1, ETV6, CEBPA) or acute lymphoblastic leukemia (RUNX1, ETV6, IKZF1, PAX5). These hereditary variants can make affected individuals more sensitive to somatic changes, facilitating the development of specific leukemia subtypes, such as those associated with ETV6 and hyperdiploid acute lymphoblastic leukemia.
In summary, predisposition syndromes to hematological neoplasms represent a unique combination of genetic and environmental factors that require multidisciplinary attention to maximize the benefit for patients.
Brain tumor
It is estimated that approximately 8% of brain tumors in children are due to inherited mutations in cancer-associated genes. Some specific types of brain tumors are linked to a higher percentage of cancer predisposition syndromes, which tend to have high penetrance. For example, rhabdoid tumors (SMARCB1) and choroid plexus carcinomas (TP53) have been associated with a high percentage of cancer predisposition.
High rates of mutations in cancer-related germline genes, such as NF2, LZTR1, SMARCB1, or SMARCE1, have been detected in cases of meningiomas and schwannomas that occur before the age of 25. In patients with medulloblastoma, approximately 6% carry an inherited mutation in a cancer-related gene. In certain specific subgroups of medulloblastoma, this rate is significantly higher, reflecting a greater genetic risk of developing cancer in these populations.
In children with medulloblastoma where the Sonic hedgehog signaling pathway is affected, those diagnosed at an older age (5 to 15 years) have a high probability of carrying a hereditary mutation in TP53, while those diagnosed at a younger age (0 to 3 years) typically present a hereditary mutation in the SUFU gene (Gorlin syndrome). Later, loss-of-function mutations in the ELP1 gene were identified in 14% of patients with medulloblastoma affected by the Sonic hedgehog pathway; ELP1 encodes a subunit of the elongator complex related to proteostasis. Additionally, variants in GPR161 were identified in rare cases of this type of medulloblastoma.
Children with WNT-induced medulloblastoma who do not present somatic variants in CTNNB1 generally carry hereditary mutations in the APC gene.
High-grade childhood glioma is linked to Li-Fraumeni syndrome and hereditary mismatch repair defect disease. In this disease, specific immunohistochemical types, specific immunological profiles, and a hypermutator phenotype suggest Li-Fraumeni syndrome. Moreover, extremely hypermutated brain gliomas have been associated with pathogenic variants in mismatch repair polymerase (POLE). In two analyzed cases, one with medulloblastoma and the other with a hypermutated high-grade glioma, these genetic links have been identified. On the other hand, low-grade glioma is associated with neurofibromatosis type 1 and sometimes with other pathologies related to the RAS activation pathway.
Blastomas
Approximately 10% of retinoblastoma cases occur in patients with germline predisposition, although specific hereditary mutations are not usually identified. The likelihood increases significantly if there is a family history of breast or ovarian cancer, as well as other types of cancer related to genital development.
Wilms tumor, also known as nephroblastoma, is considered a prime example of how abnormal processes can be related to carcinogenesis. About 10-15% of children with this tumor have a genetic cancer predisposition syndrome, although this variation may be linked to the patient’s origin.
The genes involved include WT1, TRIM28, REST, and CTR9, which are specific to Wilms tumor, as well as other genes like PALB2 or BLM. Genes like TRIM28 and REST rarely mutate somatically, indicating that their oncogenic effects are restricted to early developmental stages. The diagnosis of bilateral Wilms tumor shows a bimodal age distribution, suggesting a genetic complexity that is not fully understood. Not all patients carry known genes that predispose to its development, indicating the existence of additional genetic factors. In 12% of bilateral cases, epigenetic changes and heritable mutations affecting H19 or IGF2 have been observed. Although some cases may be explained by early somatic mosaicism, all patients with bilateral Wilms tumor should be investigated as potential carriers of cancer-predisposing genes, which could be passed on to offspring.
The suspicion of predisposition is heightened by young age, bilateral or multifocal presentation of the tumor, congenital abnormalities (especially urological), overgrowth syndromes, and specific histological types like the stromal subtype in WT1 mutants and the differentiated epithelial tumor in TRIM28 mutants. Although survival rates are high, genetic counseling and studies to assess risks are recommended, especially for siblings and future generations.
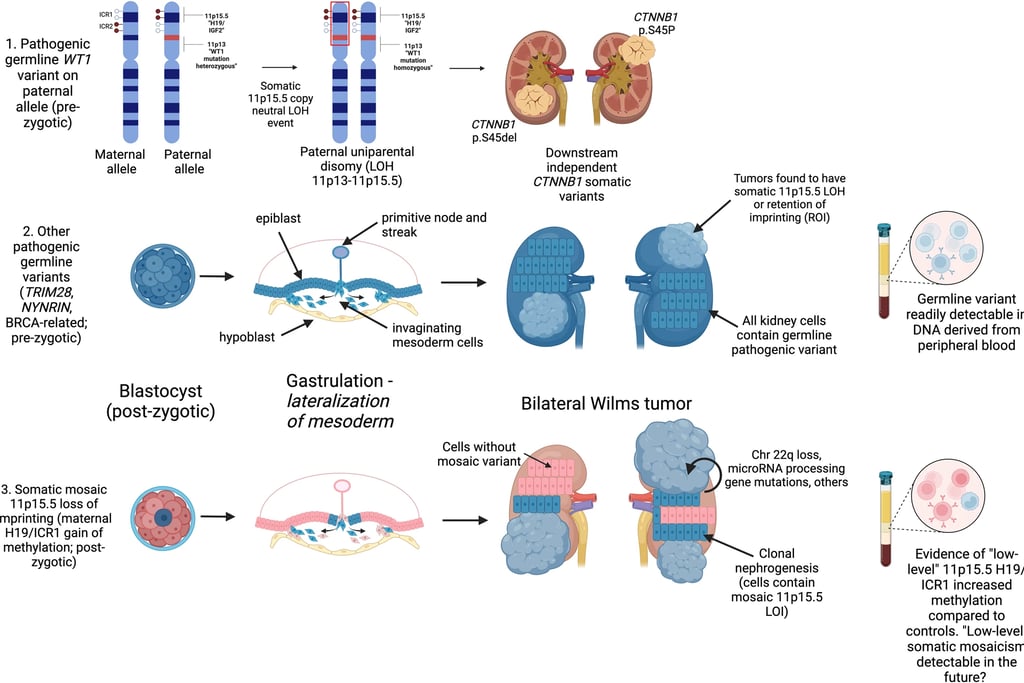
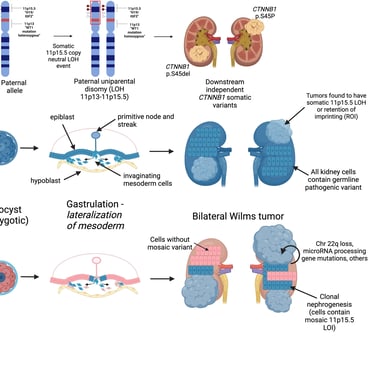
Image: Murphy et al. Genetic and epigenetic features of bilateral Wilms tumor predisposition in patients from the Children’s Oncology Group AREN18B5-Q. Nat Commun 14, 8006 (2023)
Hepatoblastoma is a rare liver tumor in childhood, representing approximately 1% of pediatric cancers. Its development is associated with genetic conditions such as Beckwith-Wiedemann syndrome and familial adenomatous polyposis, linked to mutations in the APC gene (located at locus 5q22.2). While Beckwith-Wiedemann syndrome is typically diagnosed by its clinical features, there are cases where no obvious phenotypic characteristics are observed, but epigenetic alterations in the 11p15.5 locus can be detected through molecular studies.
In children with hepatoblastoma who do not present somatic mutations in the CTNNB1 gene (locus 3p22.1), genetic testing is recommended to detect mutations in APC, as these can occur spontaneously without a family history of polyposis. Since mutations in APC and CTNNB1 are mutually exclusive, APC gene analysis may be limited to cases where no alterations in CTNNB1 are identified.
Complete DNA exome analyses of large patient groups suggest that most sporadic neuroblastomas are not associated with genetic syndromes that increase cancer risk, even in young individuals. It is important to investigate mutations in the ALK (locus 2p23.1) and PHOX2B (locus 3p11.1) genes, which are the primary causes of neuroblastoma predisposition, especially in familial cases or when cancer affects multiple areas. Associations with genetic syndromes are rare. A relationship has also been found between neuroblastoma risk and certain variants in the TP53 gene (locus 17p13.1), as well as with genetic polymorphisms that affect susceptibility. Some of these polymorphisms modify regions of DNA that act as regulators in the LMO1 gene (locus 11p15.5), which is key for controlling the overexpression of the MYCN gene (locus 2p24.3). Such variants, found in non-coding regions of DNA, illustrate how interactions between acquired genetic events and non-coding regions of the genome can influence susceptibility to high-risk neuroblastomas.
Sarcoma
Several genetic predisposition syndromes are associated with an increased risk of specific sarcomas and soft tissue tumors. Probably the most well-known association of a cancer predisposition syndrome with sarcoma is Li-Fraumeni syndrome. Osteosarcoma is the most frequent malignancy in children with Li-Fraumeni syndrome (30% of all tumors), followed by soft tissue sarcomas (23% of all tumors). Another well-known, although rare, case of osteosarcoma involves biallelic variants in DNA helicases, such as BLM (locus 15q26.1), which causes Bloom syndrome, and RECQL4 (locus 8q24.3), which causes Rothmund-Thomson syndrome.
Heterozygous variants in the RECQL4 gene (locus 8q24.3) have been shown to be significantly overrepresented in children with osteosarcoma. Osteosarcoma is a rare cancer for which the risk of developing it in childhood can be predicted through polygenic determination. Through the analysis of more than 400 single nucleotide polymorphisms associated with height achieved in childhood and adulthood, Zhang et al. developed polygenic scores that were associated both with greater height and an increased risk of osteosarcoma. This example illustrates how genome-wide association studies shed new light on old clinical observations, such as the possible relationship between taller stature and osteosarcoma.
Pathogenic (or potentially pathogenic) heterozygous germline mutations in various DNA repair genes have been shown to be enriched in a cohort of patients with Ewing sarcoma. The authors suggest that these heterozygous germline mutations may cause DNA breaks and, subsequently, fusion between the EWSR1 gene (locus 22q12.2) and genes encoding the ETS transcription factor family (EWSR1–ETS fusion), a hypothesis that may be relevant to other types of cancer and warrants further investigation. Genome-wide association studies have also highlighted the cooperation between constitutional polymorphic microsatellites and acquired EWSR1–ETS fusions. The replacement of an A with a T in the enhancer region of the EGR2 gene (locus 6q23.3) significantly affects the ability of the EWSR1–ETS chimeric proteins to bind to this regulatory region and, therefore, exert their oncogenic effects. This example again illustrates the role of the non-coding part of the genome in susceptibility to childhood cancer and may partly explain why Ewing sarcoma is more prominent in patients of European white ancestry than in other ethnic backgrounds.
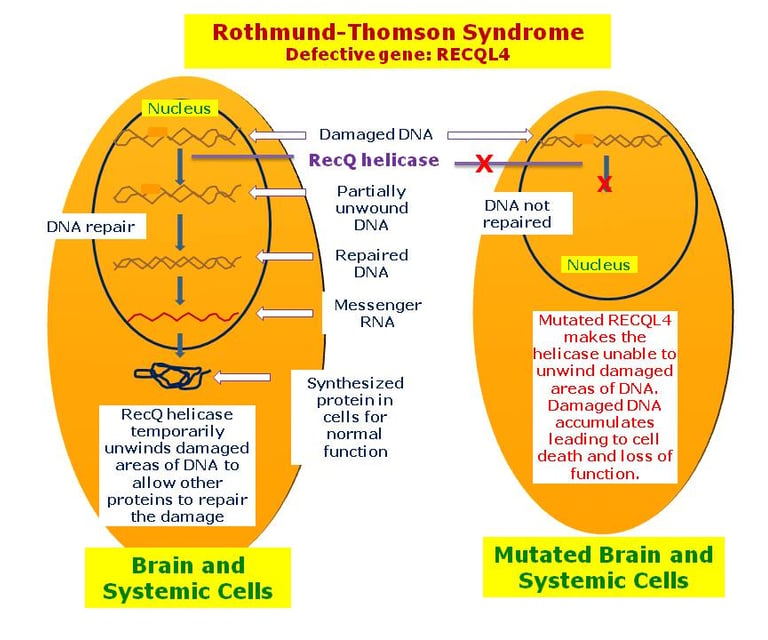
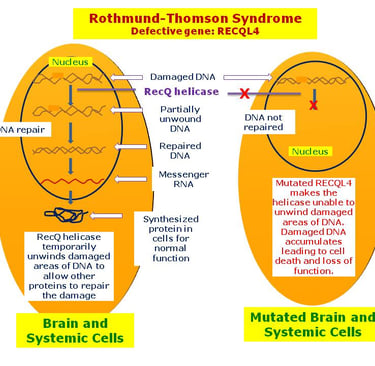
image: disorders.eyes.arizona.edu
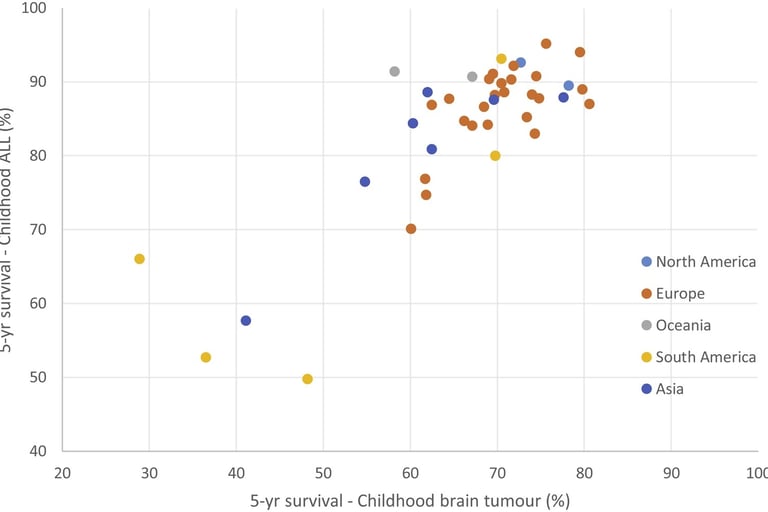
image: Erdmann et al. Childhood cancer: Survival, treatment modalities, late effects and improvements over time. Cancer Epidemiology. 2021
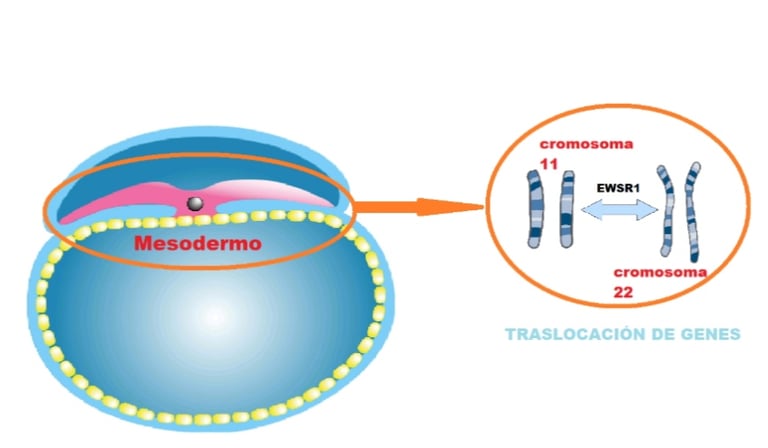
image: microbacterium.es
Treatments in Pediatric Cancer: Advances and Challenges in Precision Medicine
Recent advances in precision medicine have transformed the landscape of pediatric cancer treatment, particularly in high-risk cancer cases. The incorporation of tools commonly used in diagnosis, such as next-generation sequencing (NGS), and the development of targeted therapies have allowed for the identification of new treatment strategies. These advancements have shown considerable potential in personalizing treatments based on the molecular characteristics of each tumor. However, despite the progress, the adoption of targeted therapies remains limited, with acceptance rates ranging from 10% to 33%. This is primarily due to uncertainty about the effectiveness of these therapies and the difficulty in balancing the associated benefits and risks.
Precision Medicine and Targeted Therapies
In pediatric oncology, advances in precision medicine have enabled the identification of molecular targets in over 65% of children with high-risk cancers. Studies such as INFORM and GAIN have shown that tumor responses may be linked to specific activating genetic fusions. For example, certain gene fusions, like ALK or ROS1, are associated with better responses to targeted therapies.
Despite these advances, it is still unclear which patients will benefit the most from precision-guided therapies. Next-generation sequencing has allowed the identification of mutations in key genes, opening the door to more specific treatments. In studies like PRISM (ZERO Childhood Cancer Precision Medicine Program), molecular targets in patients' tumors were identified, and personalized treatments were offered with a follow-up of at least 18 months.
Precision-guided therapies (PGT) vs. non-guided or standard treatments: comparison of survival and treatment response
In a comparative analysis between patients treated with precision-guided therapies (PGT) and those treated with non-guided therapies (such as standard treatments), progression-free survival (PFS) outcomes were significantly better in the former. For example, patients treated with PGT showed a two-year PFS of 27%, compared to 11% in those who received non-guided treatments.
However, the difference in overall survival (OS) did not reach statistical significance in all cases, highlighting the need for further studies to assess the long-term impact of these personalized therapies.
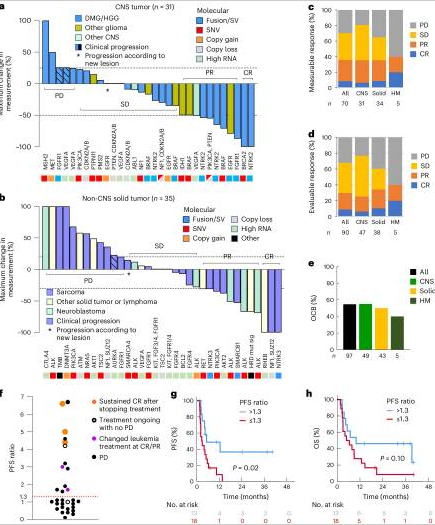
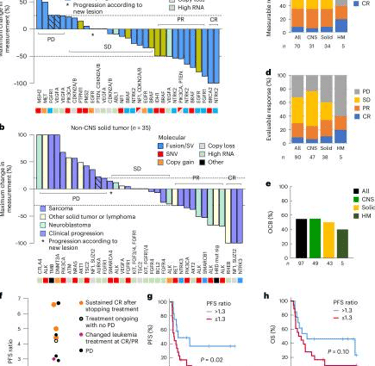
Figure: Lau LMS et al. Precision-guided treatment in high-risk pediatric cancers. Nat Med. 2024 Jul;30(7):1913-1922.
Response to Targeted Treatments: Genetic Fusions and Mutations
The success of targeted therapies varied depending on the genetic alterations present in the patients' tumors. Gene fusions and structural variations (SV) showed the highest response rates, with 60% of patients responding favorably to targeted treatments. In comparison, single nucleotide mutations (SNV) had a response rate of 32%, while other alterations, such as copy number variations (CNV), had response rates of only 14%.
For example, ALK and ROS1 fusions were key targets in patients with pediatric lung cancer and neuroblastoma, showing a notably high response rate when targeted with specific treatments.
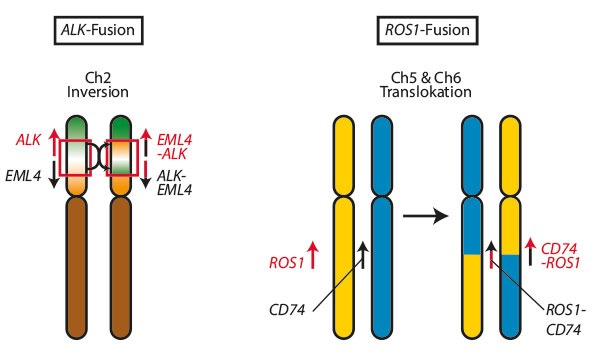
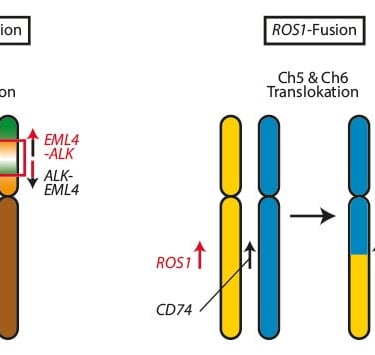
Image: Trillium.de
Challenges and Future of Targeted Therapies: Limitations in Access to Medications and Clinical Trials
Despite the promising results of precision medicine, there are significant challenges in its widespread implementation. Access to specific targeted drugs for pediatric genomic alterations and the availability of appropriate clinical trials remains limited, hindering the benefit for many patients. Greater investment is needed in the development of targeted therapies specifically for children and in the design of more inclusive clinical trials.
In summary, precision medicine has significantly improved treatment options for children with high-risk cancers, particularly those with well-established genetic alterations. However, challenges in implementing these therapies and the lack of definitive data on their impact on overall survival remain critical points. As more studies focus on genetic fusions, single nucleotide mutations, and other specific molecular changes, we are likely to see continued progress in the effectiveness of these treatments. The integration of personalized medicine in pediatric oncology is essential to improving survival rates for children with cancer.
References:
Erdmann, F. et al. Childhood cancer: Survival, treatment modalities, late effects and improvements over time. 2021. Cancer Epidemiology. 71b: 101733
Lau LMS et al.Precision-guided treatment in high-risk pediatric cancers. Nat Med. 2024 Jul;30(7):1913-1922
Kratz, Christian P et al.Predisposition to cancer in children and adolescents. The Lancet Child & Adolescent Health, Volume 5, Issue 2, 142 - 154
Nakano Y, Rabinowicz R, Malkin D. Genetic predisposition to cancers in children and adolescents. Curr Opin Pediatr. 2023 Feb 1;35(1):55-62.
Roganovic J. Genetic predisposition to childhood cancer. World J Clin Pediatr. 2024 Sep 9;13(3):95010
PDQ Cancer Genetics Editorial Board. Cancer Genetics Risk Assessment and Counseling (PDQ®): Health Professional Version. 2025 Jan 3.
Orbach D., Brech B. I. et al. The role of cancer predisposition syndrome in children and adolescents with very rare tumours. 2023. EJC Paediatric Oncology. 2: 100023
