


Hemofilia: qué es, síntomas, causas y avances en tratamiento
José Hernández Jiménez
4/17/202627 min leer


1. Introducción
La hemofilia es, en esencia, un problema en la forma en que nuestra sangre se solidifica. Por cierto, se trata de un trastorno hemorrágico hereditario que va estrechamente ligado al cromosoma X. Lo que ocurre en el organismo es que existe una deficiencia o disfunción de unas proteínas clave denominadas factores de coagulación.
Dependiendo de cuál sea la proteína que falla, los especialistas distinguen entre dos variantes: la hemofilia A, relacionada con el factor ocho, y la hemofilia B, que afecta al factor nueve. De hecho, esta alteración provoca que el cuerpo sea incapaz de formar coágulos estables. Esto se traduce en que las personas pueden sufrir hemorragias prolongadas, ya sea de forma espontánea o tras recibir traumatismos leves que, en otras circunstancias, no tendrían mayor importancia. Aunque globalmente se clasifica como una enfermedad rara, su impacto clínico es muy profundo debido a su cronicidad y a su potencial gravedad.
Desde una perspectiva biomédica, esta patología es fascinante porque se considera un modelo paradigmático de enfermedad monogénica. ¿Qué significa esto? Básicamente, que al depender de un solo gen, ha facilitado mucho el desarrollo de estrategias terapéuticas altamente dirigidas.
Sin duda, la forma de tratarla ha dado un vuelco espectacular en las últimas décadas. Hemos pasado de aplicar simplemente tratamientos paliativos para aliviar los síntomas a utilizar enfoques preventivos y, lo que es más emocionante, soluciones que son potencialmente curativas. Sin ir más lejos, la hemofilia ha sido uno de los grandes campos de prueba para el avance de la terapia génica.
La realidad actual de los pacientes ha cambiado radicalmente gracias a los saltos gigantescos en biotecnología, farmacología y lo que hoy llamamos medicina personalizada. Estos pilares han transformado por completo el pronóstico de quienes conviven con esta afección.
En particular, el desarrollo de nuevas herramientas como las terapias sustitutivas de larga duración, junto con los agentes no sustitutivos y la ya mencionada terapia génica, ha modificado de arriba abajo la calidad de vida. Ya no solo hablamos de sobrevivir, sino de una expectativa de supervivencia y un bienestar que hace años eran impensables. Al final, la ciencia ha logrado que una condición que antes era sumamente limitante se gestione hoy con una precisión asombrosa.
2. Hemofilia: qué es, síntomas y avances que están cambiando la vida de los pacientes
La visión que tenemos hoy de la hemofilia no tiene nada que ver con la de hace unas décadas. Hemos pasado de verla como una enfermedad altamente discapacitante a considerarla una condición que se puede controlar cada vez mejor.
Si miramos atrás, el panorama era bastante desolador; los pacientes se enfrentaban a una mortalidad muy elevada y a complicaciones realmente graves. Lo más temido eran, sin duda, las hemorragias intracraneales o esas hemorragias articulares recurrentes que terminaban por destrozar la movilidad. Por suerte, la introducción de la profilaxis con factores de coagulación cambió radicalmente este escenario, marcando un antes y un después en la vida de muchas personas.
Actualmente, la clave del tratamiento ya no es reaccionar al problema, sino adelantarse a él. El enfoque médico se centra totalmente en la prevención de sangrados. Esto se consigue mediante la administración constante de factores recombinantes o el uso de las llamadas terapias no sustitutivas.
Gracias a este cambio de estrategia, se ha logrado reducir de forma drástica la aparición de hemorragias espontáneas. Sin embargo, lo más valioso no es solo el dato clínico, sino cómo esto se traduce en una mejora real de la calidad de vida, permitiendo que los pacientes lleven una rutina mucho más normalizada.
Uno de los avances que más ha facilitado el día a día de los pacientes es la llegada de los tratamientos de larga duración. Al reducir la frecuencia con la que hay que administrar el medicamento, la adherencia al tratamiento es mucho mayor; es decir, es más fácil no saltarse ninguna dosis.
De hecho, la innovación no se queda ahí. Han aparecido nuevos agentes capaces de modular la coagulación, como los anticuerpos biespecíficos, que han abierto caminos que antes no existían. Esto es especialmente importante para los pacientes con inhibidores, un grupo que tradicionalmente ha tenido muchas más dificultades para encontrar un tratamiento eficaz.
Para terminar este apartado, no podemos ignorar la irrupción de la terapia génica, que ha supuesto un auténtico cambio de paradigma. Estamos hablando de algo que parecía ciencia ficción hace poco: terapias ya aprobadas que permiten que el propio organismo recupere una producción endógena y sostenida de factores de coagulación.
Lo más asombroso es que esto se logra tras una única administración. Sin duda, estamos ante un avance sin precedentes que promete transformar de manera definitiva el futuro de quienes conviven con esta patología.
3. ¿Qué ocurre en la sangre de una persona con hemofilia?
Factores de coagulación
Para empezar, conviene entender que, en condiciones normales, nuestro cuerpo realiza un proceso asombrosamente intrincado para detener cualquier pérdida de sangre. Se trata de una auténtica cascada de activación enzimática en la que intervienen múltiples factores que trabajan de forma coordinada.
De hecho, cuando todo funciona bien, esta reacción en cadena permite que la sangre pase de estado líquido a sólido justo donde se necesita. Sin embargo, en el caso de la hemofilia, este engranaje se rompe. La falta o el mal funcionamiento del factor VIII o del factor IX interrumpen este flujo de señales, lo que acaba impidiendo la formación adecuada de fibrina. Sin esta sustancia, es imposible que el cuerpo genere coágulos estables capaces de sellar una herida de manera definitiva.
Por cierto, algo que suele llevar a confusión es pensar que estas personas no dejan de sangrar nunca de forma inmediata. La realidad es que la formación inicial del tapón plaquetario —la primera respuesta del organismo— se produce sin ningún problema.
Sin embargo, el verdadero inconveniente surge justo después, ya que la falta de proteínas de coagulación compromete su estabilización. Es decir, el tapón se crea, pero no se "sujeta" bien. Como resultado, los pacientes se enfrentan a sangrados prolongados que pueden volverse un reto importante, incluso cuando se producen ante lesiones mínimas que para cualquier otra persona serían insignificantes.
Tipos A y B
Para entender esta condición, lo primero que debemos mirar es nuestra propia genética. De hecho, tanto una variante como la otra son enfermedades recesivas ligadas al cromosoma X. Esto es precisamente lo que explica que, por una cuestión de herencia, exista una mayor prevalencia en varones. Básicamente, es una peculiaridad de nuestro ADN la que determina quién tiene más probabilidades de manifestar el trastorno.
Ahora bien, aunque solemos hablar de ella de forma general, lo cierto es que existen diferencias importantes según la proteína que falte. La hemofilia A es, con diferencia, la forma más habitual y se produce por una deficiencia del factor ocho.
Por otro lado, nos encontramos con la hemofilia B, la cual está causada por la falta o el mal funcionamiento del factor nueve. A decir verdad, si nos fijamos solo en los síntomas, ambas son clínicamente similares; sin embargo, cuando entramos en el terreno de las soluciones médicas, la historia cambia bastante.
Por cierto, donde realmente se nota la distinción entre ambos tipos es en la respuesta terapéutica y en el diseño de nuevas estrategias para combatirlas. Resulta que, en el campo de la terapia génica, la investigación ha avanzado de forma desigual pero fascinante.
En este sentido, la hemofilia B ha tomado cierta ventaja tecnológica. Se ha observado que, en este tipo concreto, existe una mayor estabilidad en la expresión del factor cuando se aplican estas técnicas de vanguardia. Sin duda, este detalle es fundamental para los científicos, ya que permite que el tratamiento sea mucho más predecible y duradero en el tiempo.
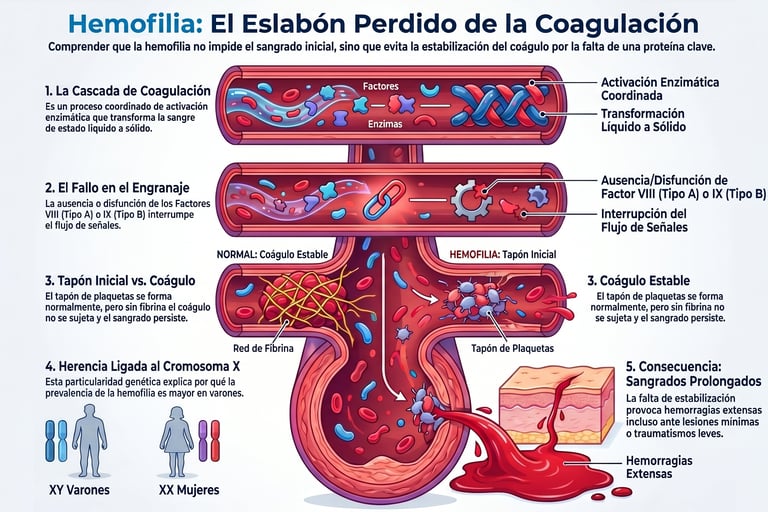
imagen: propia
4. Síntomas y riesgos reales
Hemorragias internas
Una de las formas más claras en las que se manifiesta esta condición son los sangrados que surgen sin necesidad de un golpe previo. Estas hemorragias espontáneas tienden a localizarse, sobre todo, en las articulaciones y los músculos, convirtiéndose en uno de los principales signos clínicos que debemos vigilar.
Sin embargo, el problema no termina con el sangrado en sí. Por cierto, estos episodios suelen venir acompañados de un dolor bastante molesto y una inflamación evidente en la zona afectada. Al final, todo esto termina provocando una limitación funcional, lo que dificulta que la persona pueda moverse o realizar sus actividades cotidianas con total normalidad.
Daño articular
De hecho, cuando los sangrados en las articulaciones —lo que técnicamente conocemos como hemartrosis— se repiten de forma constante, el organismo termina sufriendo las consecuencias. Esta situación desemboca inevitablemente en una artropatía hemofílica. Por cierto, no estamos hablando de un problema pasajero, sino de una degeneración progresiva de las articulaciones que va minando la salud del paciente poco a poco.
Sin embargo, lo más preocupante de este proceso es que el daño es acumulativo. Esto significa que cada episodio de sangrado deja una huella que se suma a la anterior, provocando a largo plazo un dolor crónico y una discapacidad que puede llegar a ser muy limitante.
A decir verdad, este deterioro puede dar sus primeros pasos incluso en la infancia. Por este motivo, resulta vital actuar cuanto antes; si no se instaura un tratamiento profiláctico a tiempo, el desgaste articular empezará a ganar terreno desde los primeros años de vida, condicionando el futuro de la persona.
Casos graves
Cuando entramos en los cuadros de mayor severidad, nos enfrentamos a situaciones que son realmente serias. En estos casos, pueden aparecer hemorragias intracraneales o incluso gastrointestinales, complicaciones que, a decir verdad, representan un riesgo vital inmediato para el paciente. De hecho, aunque es innegable que los tratamientos han avanzado una barbaridad, este tipo de episodios siguen siendo una preocupación clínica de primer orden que los especialistas vigilan muy de cerca.
Sin embargo, hay una realidad que no podemos ignorar: lo que tenemos hoy en día no siempre es suficiente. Resulta que, incluso cuando se sigue de forma estricta una profilaxis, hay pacientes que no dejan de sufrir sangrados recurrentes.
Esta situación es frustrante porque el daño articular sigue progresando en muchas personas, lo que deja en evidencia una clara necesidad de terapias que sean mucho más eficaces. Al final, el objetivo sigue siendo encontrar soluciones que logren frenar por completo estas complicaciones y mejoren la protección real del organismo.
5. ¿Se puede vivir con normalidad?
Deporte
Hoy en día el panorama ha cambiado radicalmente para quienes conviven con esta condición. Es genial ver cómo la mayoría de los pacientes ya se sienten con la confianza necesaria para realizar actividad física moderada de forma habitual. Al final, este movimiento constante no es solo una cuestión de estética o de estar en forma, sino que contribuye de manera directa a fortalecer la salud articular, algo que resulta vital para evitar complicaciones a largo plazo.
Por cierto, aunque mantenerse activo es fantástico, no debemos olvidar que la clave reside en el tipo de ejercicio que se elige. Por eso, lo que más se suele recomendar en estos casos son los deportes de bajo impacto, ya que permiten obtener todos los beneficios del entrenamiento sin someter al cuerpo a riesgos innecesarios. Sin embargo, más allá de la disciplina concreta que se practique, lo verdaderamente importante es que este cambio de estilo de vida mejora de forma notable la calidad de vida de las personas, aportándoles una vitalidad que se refleja en su día a día.
Infancia
Apostar por un tratamiento temprano marca una diferencia abismal en la evolución de los pacientes. De hecho, cuando se pone en marcha una profilaxis adecuada directamente durante la infancia, los resultados son realmente esperanzadores.
Este enfoque preventivo no solo ayuda a que los más pequeños crezcan con mayor seguridad, sino que es la herramienta clave para evitar el daño articular crónico que tanto condicionaba el futuro. Por cierto, gracias a esta intervención precoz, hoy es perfectamente posible favorecer un desarrollo normal, permitiendo que los niños alcancen su potencial sin las limitaciones físicas que solían ser la norma.
Calidad de vida
Es increíble cómo han cambiado las cosas. De hecho, gracias a los últimos avances terapéuticos, muchísimas personas han logrado algo que antes parecía inalcanzable: llevar una vida prácticamente normal.
Este cambio de rumbo no ha ocurrido por arte de magia. Por cierto, existen dos factores que han sido totalmente determinantes en esta evolución. Por un lado, la drástica reducción de episodios hemorrágicos ha devuelto la tranquilidad a los pacientes; por otro, la simplificación de los tratamientos ha hecho que el día a día sea mucho más llevadero. Al final, que una terapia sea más fácil de seguir cambia por completo la perspectiva de quien convive con la enfermedad.
Sin embargo, no podemos ignorar la otra cara de la moneda. A decir verdad, nos encontramos ante un obstáculo que todavía empaña estos progresos: el acceso a terapias avanzadas sigue siendo tremendamente desigual a nivel global.
No es un problema menor. Esta brecha entre países representa un desafío importante en salud pública que nos recuerda que, aunque la ciencia vuela, la equidad todavía va a paso lento. Es fundamental que estos beneficios no se queden solo en unos pocos lugares, sino que lleguen a todo aquel que los necesite, sin importar dónde viva.
6. Tratamientos actuales: De la sustitución a la modulación génica
La manera de tratar la hemofilia ha vivido una transformación radical en apenas diez años. No se trata solo de un avance más; de hecho, estamos ante un auténtico cambio de era en la medicina que ha dejado atrás viejos esquemas.
Durante mucho tiempo, nos limitamos a lo que se conocía como una estrategia puramente sustitutiva. Básicamente, el trabajo consistía en reponer aquello que al organismo le faltaba de forma externa. Sin embargo, las cosas han evolucionado muchísimo.
Por cierto, ahora nos movemos en un escenario mucho más sofisticado: un modelo multimodal y, lo que es más importante, totalmente personalizado. Ya no se busca una solución estándar para todo el mundo, sino ajustar el tratamiento a la realidad específica de cada individuo.
Hoy por hoy, el enfoque ha dejado de ser exclusivamente técnico para volverse más humano. Ya no nos conformamos simplemente con evitar el sangrado, aunque siga siendo una prioridad. De hecho, el gran objetivo actual es normalizar la vida de quien convive con esta patología.
Para alcanzar esta meta, la medicina se apoya ahora en unos pilares fundamentales que permiten al paciente mirar al futuro con otros ojos, integrando el tratamiento en su rutina de forma casi invisible. Sin duda, el progreso no solo está en los fármacos, sino en cómo estos permiten que la persona recupere el control total de su tiempo.
6.1 Terapia con factores: Optimización farmacocinética
Históricamente, la piedra angular para abordar esta condición ha consistido en el uso de concentrados, algo que a día de hoy sigue siendo fundamental. De hecho, el proceso se centra en administrar el Factor VIII a quienes conviven con la hemofilia A, mientras que para la hemofilia B el recurso esencial es el Factor IX
Sin embargo, lo que realmente ha supuesto una revolución no es solo la sustancia en sí, sino la manera de aplicarla. Por cierto, el enfoque médico ha experimentado una evolución impresionante; hemos pasado de un modelo puramente reactivo, donde solo se intervenía cuando ya existía un problema, a una estrategia de profilaxis sistemática.
A decir verdad, este cambio de mentalidad busca adelantarse a las complicaciones antes de que aparezcan, permitiendo que el tratamiento sea preventivo y constante en lugar de una simple respuesta a una urgencia.
Avances en Ingeniería Proteica
La ciencia ha avanzado muchísimo en la forma en que manejamos las proteínas de coagulación para que duren más tiempo en el organismo. De hecho, mediante técnicas muy sofisticadas como la pegilación, la fusión con fragmentos de inmunoglobulina o incluso la unión a la albúmina, se ha logrado desarrollar factores de vida media extendida. Lo que hacen estas tecnologías, básicamente, es conseguir que el medicamento permanezca activo en el cuerpo durante periodos mucho más largos de lo que era posible antes.
Este avance ha supuesto, por cierto, un alivio enorme en lo que conocemos como la carga terapéutica. Al conseguir que el factor aguante más en la sangre, se ha alcanzado una reducción en la frecuencia de infusión realmente impresionante, que oscila entre el 50% y el 80%. Es un cambio brutal para el día a día. Al no tener que pincharse tan a menudo, los niveles plasmáticos se mantienen mucho más constantes, lo que se traduce en una mayor estabilidad para el paciente y, lógicamente, favorece una mejor adherencia al tratamiento al ser mucho más sencillo de cumplir.
Sin embargo, no todo es un camino de rosas y todavía nos enfrentamos a ciertas limitaciones persistentes. A pesar de estas mejoras tan claras, sigue siendo necesaria la administración intravenosa, lo cual siempre supone una incomodidad para el paciente.
Además, existe una alta variabilidad farmacocinética entre una persona y otra; a decir verdad, el cuerpo de cada individuo procesa el fármaco de manera distinta, lo que complica la estandarización. Por si fuera poco, no podemos perder de vista el riesgo de desarrollar inhibidores, un problema que sigue siendo una preocupación clínica relevante, especialmente en los casos de hemofilia A.
6.2 Profilaxis moderna: Medicina de precisión
Hasta hace no mucho, la prevención se basaba en calendarios bastante cuadriculados, como el clásico esquema de inyectarse tres veces por semana sin importar demasiado quién fuera el paciente. Sin embargo, las cosas han cambiado radicalmente. De hecho, la profilaxis ha dejado de ser una receta fija para transformarse en una estrategia personalizada que se adapta al perfil único de cada individuo. Ya no se trata de tratar la enfermedad de forma genérica, sino de entender cómo vive y cómo reacciona cada persona.
Para que este traje a medida funcione, los médicos se fijan ahora en una serie de variables de ajuste que son fundamentales. Por cierto, ya no solo se mira el reloj; ahora se analizan con lupa los niveles valle, que es ese punto mínimo de factor que queda en la sangre antes de la siguiente dosis.
Pero la cosa no queda ahí. También se tiene muy en cuenta el fenotipo hemorrágico de cada uno y, por supuesto, el nivel de actividad física que realiza el paciente en su día a día. A decir verdad, lo más fascinante es el uso de herramientas de vanguardia, como los modelos farmacocinéticos bayesianos, que permiten predecir con una precisión asombrosa cómo se comporta el tratamiento en cada organismo concreto.
Gracias a estos avances, el listón de lo que consideramos aceptable ha subido mucho. El nuevo estándar mínimo es mantener de forma constante niveles de factor por encima del 3 al 5%.
Sin embargo, el objetivo actual va mucho más allá de ese simple número; lo que la medicina busca hoy es alcanzar una hemofilia funcional leve. La idea de fondo es que el paciente no solo esté protegido contra accidentes, sino que su cuerpo se comporte como si tuviera una variante mucho más suave de la afección.
Al final, todo este esfuerzo tecnológico tiene un impacto directo y tangible. De hecho, no solo se busca prevenir la hemartrosis y el consecuente daño articular que tanto preocupa a largo plazo.
El verdadero éxito de este modelo es conseguir la normalización completa de la actividad diaria. Se trata de que las personas puedan trabajar, estudiar y disfrutar sin el miedo constante a una crisis, logrando además una reducción drástica de las hospitalizaciones. En definitiva, el objetivo es que la condición sea solo una parte de la vida del paciente, y no algo que la condicione por completo.
6.3 Terapias no sustitutivas: La revolución farmacológica
Estamos viviendo, sin duda, el momento más disruptivo en la historia de esta patología. De hecho, la forma de abordar la enfermedad ha dado un giro total, permitiendo ahora tratamientos por vía subcutánea con intervalos prolongados, algo que mejora la rutina de los pacientes de una manera que antes ni imaginábamos.
A. Anticuerpos biespecíficos (Emicizumab)
Uno de los avances más destacados es el emicizumab. Este fármaco es fascinante porque, básicamente, se dedica a imitar la función natural del factor VIII. Su "truco" consiste en servir de puente para unir el factor IX activado y el factor X, consiguiendo así que la cascada de coagulación se restaure y vuelva a funcionar como debería.
A decir verdad, sus ventajas son impresionantes. Al tratarse de una administración subcutánea que puede espaciarse desde una vez por semana hasta una vez al mes, la carga del tratamiento se vuelve mucho más ligera. Además, ha demostrado una alta eficacia incluso en aquellos pacientes que presentan inhibidores, lo que supone un alivio enorme para casos que antes eran muy complejos de manejar.
B. Terapias de reequilibrio hemostático
Sin embargo, la medicina no solo se ha centrado en sustituir lo que falta. Existe otra estrategia muy inteligente: las terapias de reequilibrio hemostático. En este caso, no se reemplaza el factor, sino que se busca inhibir los anticoagulantes naturales del cuerpo para potenciar la generación de trombina.
En este campo, los blancos terapéuticos principales son la antitrombina y el inhibidor de la vía del factor tisular. Por cierto, en este año 2026, los nuevos inhibidores de la antitrombina bimensuales están dando mucho que hablar. Han logrado reducciones de hasta el 90% en sangrados, consolidándose como una alternativa robusta y fiable para ambos tipos de la enfermedad.
C. Moléculas emergentes
Si miramos hacia lo que está ocurriendo entre 2025 y 2026, la investigación actual está centrada en las llamadas moléculas emergentes. Es un campo vibrante donde se están desarrollando miméticos avanzados del factor VIII y plataformas innovadoras de ácido ribonucleico de interferencia.
Por otro lado, están cobrando mucha fuerza las terapias combinadas. Estas están diseñadas con un objetivo doble: por un lado, minimizar la inmunogenicidad (es decir, que el cuerpo no reaccione contra el tratamiento) y, por otro, reducir al máximo esos eventos hemorrágicos residuales que todavía pueden aparecer. Al final, se trata de no dejar ningún cabo suelto en la protección del paciente.
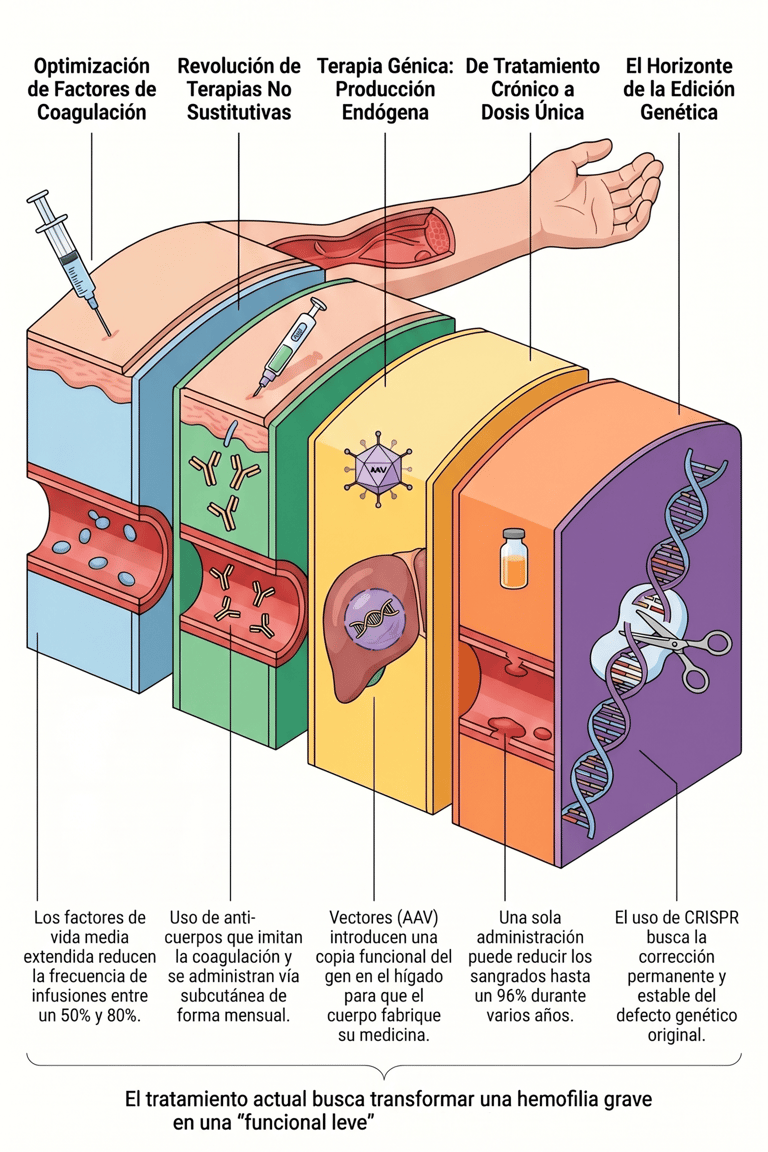
imagen: propia
7. La terapia génica: Hacia un tratamiento curativo
Estamos ante el avance más disruptivo y revolucionario que se ha visto jamás en toda la historia del tratamiento de esta enfermedad. La terapia génica no es simplemente un paso más en el camino; de hecho, supone un cambio total de escenario porque, a diferencia de los métodos tradicionales, esta estrategia se encarga de abordar directamente la causa genética del problema.
Gracias a que este enfoque va directo a la raíz de la situación, nos permite dejar atrás, por fin, ese modelo de tratamiento continuo que tanto condiciona el día a día de las personas. Sin embargo, lo más emocionante es que ahora el objetivo se centra en una intervención duradera. A decir verdad, estamos ante la posibilidad real de una opción potencialmente curativa, algo que transforma por completo las expectativas de futuro de los pacientes.
7.1 Fundamento biológico y racional terapéutico
A decir verdad, esta condición se considera el escenario perfecto para aplicar la terapia génica por varios motivos que facilitan mucho el trabajo de los científicos. El primero de ellos, y quizás el más relevante, es que estamos ante una enfermedad monogénica. Esto significa, básicamente, que el problema se origina por defectos en puntos muy específicos: los genes del factor ocho o el factor nueve. Al tener un "culpable" tan bien localizado, diseñar una solución resulta mucho más viable que en otras patologías más complejas.
El éxito de esta técnica también depende de a dónde tengamos que dirigirnos. En este caso, el órgano diana es el hígado, el cual afortunadamente es fácilmente accesible para los vectores que transportan la corrección necesaria.
De hecho, una de las grandes ventajas es que no necesitamos que la información genética se active en todos los rincones del cuerpo. Resulta que la producción de estas proteínas exclusivamente en la zona hepática es fisiológicamente adecuada para que el organismo funcione correctamente. No hace falta complicar el proceso buscando que el gen se exprese en múltiples tejidos; con que el hígado haga su parte, el objetivo está cumplido.
Sin embargo, lo que realmente hace que esta terapia sea una esperanza tan real es que no buscamos la perfección absoluta para cambiarle la vida a alguien. En los casos más complicados, los que conocemos como fenotipo severo, no hace falta alcanzar niveles de factor del cien por cien para notar una mejoría radical.
A decir verdad, con lograr apenas un pequeño aumento que nos sitúe en un cinco por ciento o algo más, ya se genera un impacto clínico impresionante. Es fascinante cómo una cifra que parece pequeña sobre el papel puede suponer el paso de una vida llena de limitaciones a una rutina mucho más libre y segura.
7.2 Tecnología de vectores AAV
La gran mayoría de las tácticas que empleamos hoy en día se apoyan en el uso de los vectores de virus adenoasociados. Estas herramientas funcionan, en esencia, como un sofisticado sistema de transporte biológico. Su misión principal es transportar e introducen una copia funcional del gen directamente en el corazón de los hepatocitos, que son las células del hígado encargadas de esta tarea.
Por cierto, lo que resulta verdaderamente innovador es el resultado de este proceso. Al conseguir que el gen se asiente correctamente, el propio organismo del paciente recupera su capacidad de trabajo. Esto permite que se genere una producción endógena y sostenida del factor de coagulación, lo que significa que el cuerpo empieza a fabricar de nuevo su propia medicina de forma constante y autónoma.
Una de las cuestiones que más suele interesar es cómo interactúa este material con nuestra propia herencia biológica. A decir verdad, el sistema es bastante ingenioso y seguro. Estos vectores generalmente no integran su ADN en el genoma del paciente, lo que evita alterar nuestra estructura genética básica.
En lugar de mezclarse de forma permanente, los genes transferidos se mantienen de forma episomal. Es decir, el material genético nuevo permanece independiente dentro de la célula, funcionando a pleno rendimiento pero sin "coserse" a nuestro ADN original. Al final, se logra un equilibrio perfecto entre eficacia y respeto a la integridad celular.
7.3 Evidencia clínica reciente
a) Hemofilia B: El mayor éxito terapéutico
Dentro de las innovaciones más punteras, la opción conocida como etranacogene dezaparvovec se ha posicionado como la alternativa más evolucionada, ofreciendo hasta ahora resultados sumamente sólidos y consistentes. De hecho, su impacto en la calidad de vida es tan notable que ha logrado una reducción drástica de episodios hemorrágicos, situándose esta mejoría clínica entre el 90% y el 96%.
Sin embargo, de nada serviría un cambio tan brusco si no fuera duradero en el tiempo. A decir verdad, esta terapia ha demostrado una durabilidad envidiable, manteniendo una expresión estable y sostenida del factor durante, al menos, un periodo de 4 a 5 años. Al final, lo que se ha conseguido es que esta mejoría no sea solo un destello pasajero, sino una nueva realidad constante para quienes conviven con esta condición.
b) Hemofilia A: Desafíos y variabilidad
Es cierto que ya disponemos de tratamientos pioneros como el valoctocogene roxaparvovec, pero no podemos ignorar que la hemofilia A nos plantea retos técnicos bastante más espinosos que otras variantes.
Para empezar, el gen del factor VIII es considerablemente más grande y, sobre todo, posee una complejidad estructural que dificulta su manejo. No es un gen sencillo de transportar o integrar, y esa naturaleza intrincada complica mucho los procesos terapéuticos.
Por cierto, otro factor que nos mantiene alerta es la alta variabilidad interindividual que vemos en la consulta. Básicamente, no todos los pacientes responden de la misma manera; mientras que algunos muestran resultados excelentes, en otros la reacción es mucho más modesta.
Además, nos enfrentamos a una realidad un tanto frustrante: en un grupo de personas, se ha observado un descenso progresivo de los niveles de factor VIII conforme pasa el tiempo tras la intervención. Esto nos indica que el efecto, por desgracia, no siempre es tan permanente como nos gustaría.
No podemos pasar por alto las complicaciones relacionadas con la tolerancia del cuerpo. A decir verdad, esta modalidad presenta una mayor inmunogenicidad, lo que significa que el sistema inmune tiende a reaccionar más contra la terapia.
A esto se le suma una mayor toxicidad hepática si comparamos los datos con otros enfoques. Son obstáculos importantes, sin duda, pero entender estos riesgos es el paso previo necesario para poder superarlos en el futuro.
7.4 Limitaciones y desafíos actuales
Es innegable que esta opción terapéutica tiene un potencial realmente revolucionario; sin embargo, no podemos ignorar que todavía existen barreras críticas que nos impiden ofrecerla a todo el mundo de forma universal.
Para empezar, nuestro propio sistema de defensa suele ser el primer desafío. Muchos pacientes presentan anticuerpos preexistentes frente a los virus que utilizamos como transporte, lo que anula su eficacia. Por si fuera poco, tras la infusión, corremos el riesgo de que se produzca una activación inmune hepática, una respuesta inflamatoria que complica mucho el proceso y obliga a los médicos a extremar las precauciones.
Otro punto delicado es la tremenda variabilidad en la respuesta de cada persona. Mientras que hay pacientes que apenas muestran reacción al tratamiento, otros experimentan un aumento tan marcado que alcanzan niveles supranormales de factor. Esta subida excesiva genera un riesgo teórico de trombosis que nos obliga a monitorizar cada caso con lupa, ya que no existe una forma estándar de predecir quién tendrá una respuesta moderada y quién una demasiado intensa.
A día de hoy, nos vemos obligados a imponer ciertas restricciones de población bastante estrictas. Por motivos de seguridad, esta terapia todavía no está indicada para el sector infantil ni para aquellos pacientes que ya tienen una enfermedad hepática previa, ya que el hígado no toleraría bien la intervención.
Finalmente, debemos hablar de la realidad logística. Se trata de una de las terapias más caras que existen a nivel global, con costes que ascienden a millones por persona. Además, no es un tratamiento que pueda aplicarse en cualquier hospital; requiere de centros altamente especializados con una infraestructura y un equipo médico preparado para manejar una tecnología de tal magnitud.
7.5 El futuro de la terapia
A día de hoy, la ciencia no se queda de brazos cruzados; al contrario, los investigadores están volcados en superar los límites que todavía encontramos en la medicina actual. Lo cierto es que el foco se ha desplazado hacia soluciones mucho más profundas y sofisticadas.
Uno de los caminos más prometedores en los que se trabaja actualmente es la edición genética, especialmente mediante el uso de la tecnología de repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas junto con sus proteínas asociadas. A decir verdad, lo que se busca con esta técnica de precisión es realizar una corrección permanente y totalmente estable del gen defectuoso. De hecho, ya no se trata solo de compensar la falta de factor, sino de arreglar el problema directamente en la raíz de nuestro mapa biológico.
Por otro lado, se está poniendo mucho empeño en la creación de nuevos vectores. La idea principal es desarrollar virus de transporte que sean mucho menos inmunogénicos, es decir, que pasen desapercibidos ante las defensas del organismo. Por cierto, conseguir que el cuerpo no reaccione de forma agresiva ante estos mensajeros es fundamental, ya que abriría la puerta a la re-dosificación. Esto permitiría, si fuera necesario, repetir el tratamiento en el futuro, algo que con las herramientas actuales resulta muy complicado debido al rechazo inmunológico.
Finalmente, otra línea de investigación fascinante se centra en la optimización de los resultados mediante el uso de reguladores de promotor. Sin embargo, no basta con que el cuerpo empiece a fabricar la proteína; lo verdaderamente importante es tener un control más fino sobre cuánta cantidad se produce. Gracias a estos reguladores, los médicos esperan poder ajustar de forma muy precisa los niveles de factor en la sangre, logrando que la producción sea constante y, sobre todo, segura para cada paciente de manera individualizada.
8. Mitos y verdades sobre la hemofilia
A veces, la imagen que tenemos de ciertas condiciones patológicas se queda anclada en el pasado, alimentando temores que ya no tienen razón de ser. Sin embargo, gran parte de lo que se suele escuchar sobre esta enfermedad responde a una percepción desactualizada que no tiene en cuenta los increíbles avances recientes en el tratamiento.
Por ejemplo, todavía existe el mito de que estas personas "no pueden hacer ejercicio" bajo ninguna circunstancia. Pero lo cierto es que eso es totalmente falso. De hecho, siempre que se cuente con un tratamiento adecuado, la práctica de ejercicio controlado no solo es posible, sino que resulta sumamente beneficioso y recomendado para mantener el bienestar físico.
Otro temor muy extendido es la idea de que este diagnóstico es siempre mortal. De nuevo, estamos ante una creencia que es rotundamente falso. Por cierto, gracias a la medicina moderna y al seguimiento de un tratamiento adecuado, la esperanza de vida actual ha mejorado tanto que puede llegar a ser muy cercana a la de la población general. Al final, lo que antes se veía con pesimismo, hoy se afronta con una normalidad que hace apenas unas décadas parecía inalcanzable.
9. Conclusión
Es asombroso ver cómo ha cambiado el panorama para quienes conviven con esta patología. Si echamos la vista atrás apenas unas décadas, nos daremos cuenta de que el manejo de la enfermedad ha vivido una transformación radical. De hecho, lo que antes se consideraba una condición altamente incapacitante y con una preocupante tasa de mortalidad, hoy se ha convertido en una condición crónica que se puede controlar perfectamente.
Este cambio de rumbo no ha sido casualidad, sino el resultado directo de impresionantes avances terapéuticos. La llegada de la profilaxis y el desarrollo de nuevos fármacos han marcado un antes y un después en la rutina de los pacientes.
Sin embargo, si hay algo que está rompiendo moldes, es la terapia génica. Por cierto, gracias a estas innovaciones, la calidad de vida ha dado un salto cualitativo enorme. Ahora es posible reducir drásticamente la frecuencia de sangrados, lo que a su vez ayuda a prevenir el daño articular crónico y, por supuesto, permite que la esperanza de vida siga aumentando de forma constante.
No todo el camino está libre de obstáculos. A pesar de estos logros, todavía nos enfrentamos a desafíos de gran calado que no podemos ignorar. Uno de los problemas más evidentes es el acceso desigual a estos tratamientos de última generación; no todos los pacientes llegan a ellos de la misma manera.
Además, los investigadores están trabajando a contrarreloj para mejorar la durabilidad de los efectos en las intervenciones genéticas y para pulir al máximo la seguridad de los procedimientos. Al final, se trata de que los beneficios no solo sean potentes, sino que se mantengan estables a lo largo de toda la vida del paciente.
Si miramos hacia lo que viene, el futuro se antoja apasionante. De hecho, ya se vislumbra un escenario donde la suma de la terapia génica, la edición genética y la medicina personalizada podría cambiarlo todo de nuevo.
Estamos ante la posibilidad real de alcanzar una cura definitiva, lo que terminaría de consolidar este histórico cambio de paradigma en el tratamiento. Ya no hablamos solo de gestionar síntomas, sino de corregir la base del problema para que la enfermedad sea, algún día, cosa del pasado.

10. Referencias
Araujo, I. de O., Suassuna, L. F., Dos Santos, I. L., & Rodrigues, D. de O. W. (2025). Efficacy, safety and satisfaction of using emicizumab in hemophilia A patients without factor VIII inhibitors: A systematic review. Hematology, Transfusion and Cell Therapy, 47(3), 103849.
Bala, N. S., & Thornburg, C. D. (2025). Gene therapy in hemophilia A: Achievements, challenges, and perspectives. Seminars in Thrombosis and Hemostasis, 51(1), 28–40.
Chen, E. C., et al. (2023). Emicizumab acquired hemophilia. Haemophilia, 29(1), 84–89.
Chen, M., Lin, Y., He, G., Huang, L., Han, J., & Ni, J. (2025). Cost-effectiveness of emicizumab for the treatment of hemophilia A: A systematic review. Frontiers in Public Health, 13, 1658760.
Deshpande, S. R., & Tong, J. (2024). Adeno-associated virus–based gene therapy for hemophilia A and B: A systematic review and meta-analysis. Blood Advances, 8(23), 5957–5969.
Forsyth, A. L., et al. (2022). Health outcomes in hemophilia: Impact of prophylaxis. Journal of Thrombosis and Haemostasis, 20(5), 1120–1130.
Huang, K. (2025). Transforming hemophilia care: Emerging therapeutic innovations and challenges. Expert Review of Hematology, 18(10), 785–802.
Jiang, D., et al. (2025). Clinical trials update in hemophilia gene therapy. Journal of Thrombosis and Haemostasis.
Kaczmarek, R., Miesbach, W., Ozelo, M. C., & Chowdary, P. (2024). Current and emerging gene therapies for haemophilia A and B. Haemophilia, 30(S3), 12–20.
Khair, K., et al. (2023). Quality of life in people with hemophilia: Current perspectives. Haemophilia, 29(1), 10–19.
Lenting, P. J., & Fong, S. (2024). Gene therapy for hemophilia. Blood Advances, 8(23), 6081–6090.
Mahlangu, J., et al. (2023a). Emicizumab prophylaxis in hemophilia A. New England Journal of Medicine, 388(2), 123–134.
Mahlangu, J., et al. (2023b). Non-factor therapies in hemophilia management. The Lancet, 401(10385), 1423–1435.
Mahlangu, J., et al. (2024). Non-replacement therapy review. Haemophilia, 30(2), 123–135.
Mannucci, P. M., & Tuddenham, E. G. D. (2023). The hemophilias—from royal genes to gene therapy. New England Journal of Medicine, 389(3), 215–225.
Miesbach, W. (2024). Advances in hemophilia treatment: Factor replacement and beyond. Haemophilia, 30(S1), 5–12.
Miesbach, W., et al. (2024). Gene therapy for hemophilia: Clinical outcomes and challenges. Blood Reviews, 58, 101045.
Mihaila, R. G. (2023). Bispecific antibody to gene therapy. Biomedical Papers, 167(1), 1–8.
Nathwani, A. C. (2022). Gene therapy for hemophilia. Hematology: American Society of Hematology Education Program, 2022(1), 569–578.
Nathwani, A. C., et al. (2023). Long-term safety and efficacy of gene therapy in hemophilia B. The Lancet Haematology, 10(2), e102–e112.
Oldenburg, J., & Mahlangu, J. N. (2023). Hemophilia: Advances in pathophysiology and treatment. Nature Reviews Disease Primers, 9(1), 15.
Oldenburg, J. (2024). Emerging therapies hemophilia. Nature Reviews Drug Discovery, 23(2), 89–104.
Oldenburg, J., et al. (2024). Current and emerging therapies for hemophilia. Nature Reviews Disease Primers, 10(1), 22.
Pang, X., et al. (2025). Advances in AAV-mediated gene therapy for hemophilia. Molecular Therapy.
Pasi, K. J., et al. (2022). Multiyear follow-up of AAV gene therapy for hemophilia. New England Journal of Medicine, 386(1), 29–40.
Pasi, K. J., et al. (2024). Targeting antithrombin and TFPI in hemophilia. Journal of Thrombosis and Haemostasis, 22(4), 567–578.
Peyvandi, F., Garagiola, I., & Young, G. (2022). The past and future of hemophilia: Diagnosis, treatments, and its complications. The Lancet, 400(10360), 1907–1920.
Pfrepper, C., Klamroth, R., Oldenburg, J., et al. (2024). Emicizumab for the treatment of acquired hemophilia A: Consensus recommendations from the GTH-AHA working group. Hamostaseologie, 44(6), 466–471.
Pipe, S. W. (2023). Hemophilia: Current management and emerging therapies. Blood, 141(20), 2350–2362.
Pipe, S. W. (2024a). Gene therapy advances. Blood, 143(10), 1023–1035.
Pipe, S. W. (2024b). Factor VIII mimetics. Blood Advances, 8(3), 789–800.
Pipe, S. W., et al. (2024c). Clinical advances in hemophilia: A review of emerging therapies. Journal of Thrombosis and Haemostasis, 22(1), 15–28.
Pipe, S. W., & Young, G. (2024d). Advances in hemophilia therapeutics. Blood, 144(2), 150–162.
Pipe, S. W. (2026). Gene therapy in hemophilia: Current status and future directions. New England Journal of Medicine, 394(4).
Raheja, P., et al. (2025). Long-term outcomes of gene therapy in hemophilia B. Haemophilia.
Shapiro, A. D., et al. (2023). Emerging rebalancing therapies in hemophilia. Blood Reviews, 57, 100987.
Skinner, M. W. (2022). Global burden hemophilia. Haemophilia, 28(3), 345–352.
Soroka, A. B., Feoktistova, S. G., Mityaeva, O. N., & Volchkov, P. Y. (2023). Gene therapy approaches for the treatment of hemophilia B. International Journal of Molecular Sciences, 24(13), 10766.
Soucie, J. M., et al. (2022). Epidemiology hemophilia. American Journal of Hematology, 97(5), 678–686.
Srivastava, A., et al. (2022). WFH guidelines for the management of hemophilia. Haemophilia, 28(Suppl 4), 1–158.
Thornburg, C. D., et al. (2023). Pediatric hemophilia management. Pediatrics, 151(4), e2022056789.
Thornburg, C. D., et al. (2025). Emerging gene therapies for hemophilia: Clinical perspectives. The Lancet Haematology.
van Vulpen, L. F. D., et al. (2022). Joint disease in hemophilia: Pathophysiology and management. Blood Reviews, 52, 100915.
VandenDriessche, T., et al. (2025). Genome editing strategies for hemophilia. Nature Reviews Drug Discovery.
Xue, F., et al. (2025). Factor IX gene therapy clinical outcomes. Blood Advances.
Yamaguti-Hayakawa, G. G., & Ozelo, M. C. (2022). Gene therapy for hemophilia: Looking beyond factor expression. Experimental Biology and Medicine, 247(24), 2223–2232.
Yang, W., et al. (2025). Meta-analysis of AAV gene therapy in hemophilia. Frontiers in Medicine.
Young, G. (2023). New developments in hemophilia treatment. Blood Advances, 7(15), 3450–3460.
Young, G., Pipe, S. W., Kenet, G., et al. (2024). Emicizumab is well tolerated and effective in people with congenital hemophilia A regardless of age, severity of disease, or inhibitor status: A scoping review. Research and Practice in Thrombosis and Haemostasis, 8(4), 102415.