


Síndrome de Edwards: una mirada actual a sus bases genéticas y retos clínicos
José Hernández Jiménez
3/18/202621 min leer


1. Introducción
El Síndrome de Edwards representa una de las realidades más complejas de la genética humana. De hecho, después del conocido síndrome de Down, esta es la trisomía que se presenta con mayor frecuencia en nuestra especie. Básicamente, el origen de todo radica en una alteración celular: la presencia de una copia adicional del cromosoma 18.
Esta carga genética extra no es un detalle menor, ya que desencadena un impacto sistémico que afecta a casi todo el organismo. Por cierto, aunque el descubrimiento de este patrón clínico se realizó hace ya varias décadas, ha sido el desarrollo reciente de la citogenética y la biología molecular lo que realmente nos ha permitido comprender la enorme heterogeneidad clínica que define a esta condición.
Lo que ocurre a nivel físico es, a decir verdad, muy delicado. Al existir ese material genético de más, el desarrollo del bebé se ve comprometido por una gran cantidad de anomalías congénitas. Es muy habitual que aparezcan anomalías cardíacas severas y diversas alteraciones neurológicas, además de un evidente retraso del crecimiento intrauterino.
Sin embargo, lo más impactante es la enorme variedad de síntomas que pueden manifestarse. De hecho, se han llegado a identificar más de un centenar de malformaciones asociadas, lo que se traduce en una elevada morbilidad y un riesgo muy alto de mortalidad tanto antes del nacimiento como en los primeros días de vida. Cada caso es un mundo, pero la fragilidad de estos pacientes es una constante que marca su pronóstico.
Afortunadamente, la medicina ha dado pasos agigantados en lo que respecta a la identificación temprana. Hoy en día, gracias a técnicas como el cribado bioquímico y la ecografía fetal, los especialistas pueden detectar indicios con mucha antelación. Por otro lado, la llegada de las pruebas genéticas no invasivas ha supuesto una auténtica revolución, permitiendo obtener información valiosa sin poner en riesgo el embarazo.
A pesar de estos avances técnicos, lo cierto es que la situación sigue planteando desafíos que van más allá de lo médico. De hecho, la detección temprana ha abierto profundos debates éticos sobre el manejo de los recién nacidos y las alternativas terapéuticas que se deben ofrecer. Al final, no se trata solo de tecnología, sino de cómo gestionar de la mejor manera posible una situación clínica tan compleja y humana.
2. La genética detrás del síndrome
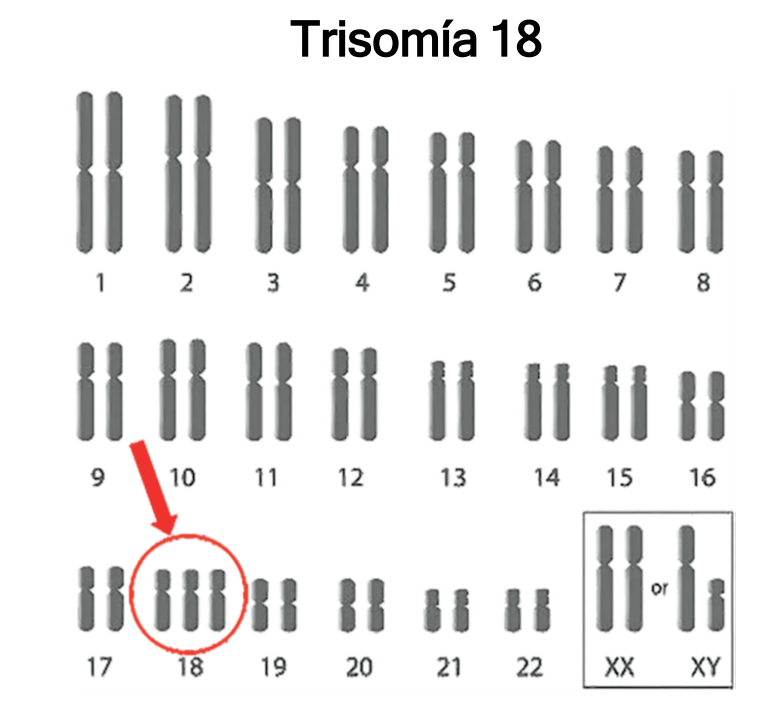
imagen: cdph.ca.gov
Para entender realmente qué ocurre en este proceso, primero debemos echar un vistazo a cómo se organiza nuestro material biológico. Lo cierto es que, en condiciones normales, nuestras células contienen cuarenta y seis cromosomas que se agrupan ordenadamente en veintitrés parejas.
Sin embargo, cuando hablamos de la trisomía 18, nos encontramos ante lo que los expertos denominan una aneuploidía. Básicamente, esto significa que existe un número anómalo de estas estructuras; de hecho, en lugar del par habitual, aparecen tres copias del cromosoma dieciocho. Ese pequeño "extra" eleva el total a cuarenta y siete y, a decir verdad, es lo que acaba alterando profundamente todo el desarrollo embrionario.
¿Cómo sucede esto exactamente? Por lo general, todo empieza con un error durante la meiosis, que es el proceso de división que da lugar a las células reproductivas. Este fallo se conoce técnicamente como no disyunción y suele ocurrir con mayor frecuencia durante la formación de los gametos maternos.
Lo que pasa es que el óvulo, por un error en la separación, acaba portando dos copias del cromosoma en cuestión. Por cierto, una vez que se produce la fecundación, ese embrión resultante termina heredando esa tercera copia adicional, marcando así el inicio de la enfermedad.
Es importante destacar que no todos los casos se manifiestan de la misma forma, ya que existen tres variantes principales desde el punto de vista de la citogenética. La más habitual, y lamentablemente la más grave, es la trisomía 18 completa. En este escenario, el cromosoma extra está presente en absolutamente todas las células del cuerpo, lo que suele traducirse en una menor supervivencia.
Por otro lado, existe la trisomía 18 parcial. Aquí no sobra un cromosoma entero, sino que solo se ha duplicado una porción del mismo. En estos casos, la situación cambia mucho; de hecho, la gravedad de los síntomas depende totalmente de qué segmento genético esté implicado.
Finalmente, nos encontramos con el patrón de trisomía 18 en mosaico. A diferencia de los anteriores, este error surge después de la fecundación, justo en las primeras divisiones de las células. El resultado es curioso: el organismo acaba siendo una mezcla donde algunas células son totalmente normales y otras presentan la alteración. Esto suele generar una expresión de la enfermedad mucho más leve o, al menos, bastante heterogénea.
Hoy en día, la ciencia nos permite identificar estas variantes con una claridad asombrosa. Para lograrlo, los especialistas utilizan técnicas de análisis muy avanzadas como el cariotipo o la hibridación fluorescente in situ, que permiten ver directamente qué ocurre en el núcleo celular.
También se emplean los microarrays cromosómicos, una tecnología punta que ayuda a comprender mucho mejor cómo se relaciona la carga genética con los síntomas que presenta cada paciente. Al final, se trata de utilizar la mejor tecnología disponible para poner nombre y apellidos a cada caso particular.
3. Qué ocurre en el organismo
Contar con una copia extra del cromosoma 18 no es un detalle menor; de hecho, este hecho desajusta por completo la expresión de numerosos genes que son vitales mientras el bebé se está formando en el útero. Esta alteración genética se entromete en procesos tan básicos como la diferenciación celular y la organogénesis, lo que acaba frenando el crecimiento fetal y provocando fallos en múltiples órganos al mismo tiempo. Básicamente, la presencia de ese material genético adicional impide que el cuerpo siga su "manual de instrucciones" original.
Uno de los puntos más críticos en esta condición es, sin duda, el sistema cardiovascular. Por cierto, una gran mayoría de los pacientes nacen con lo que los médicos llaman cardiopatías congénitas. Estamos hablando de problemas serios, como los defectos del tabique ventricular o los defectos del tabique auricular, además de la persistencia del conducto arterioso y otras malformaciones cardiacas todavía más complejas. Lamentablemente, estas complicaciones son las que más pesan en la elevada mortalidad neonatal que caracteriza a esta patología.
Pero el impacto no se detiene en el corazón. Las alteraciones neurológicas también aparecen con mucha frecuencia, ya que la embriogénesis del cerebro se ve interrumpida desde muy temprano. Esto suele manifestarse a través de anomalías estructurales del sistema nervioso central y una microcefalia evidente. Al final, todo esto se traduce en un marcado retraso en el desarrollo neurológico del niño.
Durante el embarazo, las señales suelen ser bastante claras para los especialistas. De hecho, a través de la ecografía prenatal, es habitual detectar una notable restricción del crecimiento intrauterino. Los hallazgos visuales suelen ser muy variados: desde anomalías craneofaciales y malformaciones en las extremidades, hasta fallos visibles en el sistema genitourinario o defectos en el área gastrointestinal.
Sin embargo, la lista de posibles complicaciones es todavía más larga porque la enfermedad es sistémica por naturaleza. Se han documentado casos con atresia esofágica, hernia diafragmática e incluso defectos del tubo neural. Básicamente, casi cualquier órgano del cuerpo puede verse afectado por esta carga genética extra.
Esta enorme combinación de malformaciones es lo que explica la extrema complejidad clínica del síndrome. A decir verdad, es precisamente esa afectación de tantos sistemas a la vez lo que hace que sea tan difícil establecer estrategias terapéuticas que resulten realmente eficaces para estos pacientes.
4. Síntomas y características clínicas
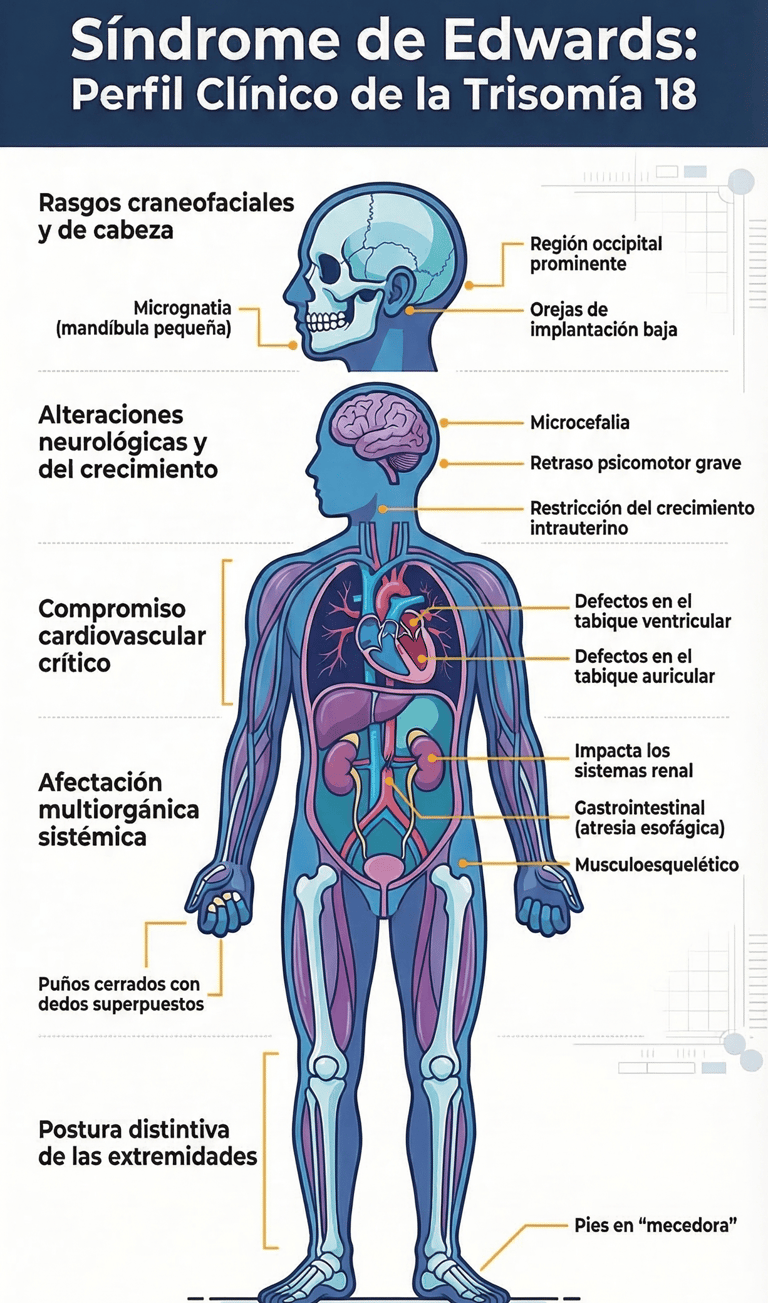
imagen: elaboración propia
El Síndrome de Edwards deja una huella física muy característica que, a menudo, permite a los médicos sospechar el diagnóstico clínico nada más nacer el bebé. Durante el periodo neonatal, es habitual observar ciertos rasgos que, en conjunto, dibujan un patrón muy específico. Por ejemplo, es frecuente que se presente un bajo peso al nacer, acompañado de una mandíbula inusualmente pequeña, lo que técnicamente conocemos como micrognatia.
A esto se le suma una región occipital del cráneo bastante prominente y unas orejas de implantación baja. Sin embargo, más allá de estos rasgos faciales, hay un signo que destaca sobre los demás por ser sumamente distintivo: la posición de las manos. De hecho, es muy habitual encontrar los puños cerrados con los dedos superpuestos, una postura que suele llamar inmediatamente la atención de los especialistas.
Por cierto, el impacto de esta trisomía 18 no se limita solo al rostro o las manos. Al observar las extremidades inferiores, es común detectar lo que se denomina pies en mecedora, además de una marcada hipoplasia de las uñas y diversas deformidades esqueléticas. Todas estas características forman parte de un patrón dismórfico que evidencia la complejidad de la alteración genética.
No obstante, lo que ocurre en el interior del organismo es lo que realmente compromete la estabilidad del paciente. Por lo general, estos niños enfrentan serias dificultades respiratorias y notables problemas de alimentación desde sus primeros días. A esto hay que añadirle un retraso grave del desarrollo psicomotor, lo que conlleva que la gran mayoría de los casos requieran de una atención médica intensiva y constante.
Sin duda, uno de los hallazgos más críticos y que más peso tiene en el pronóstico son las malformaciones cardíacas. Estas anomalías en el corazón son extremadamente frecuentes y pueden derivar en situaciones de insuficiencia cardíaca o hipoxemia. Al final, este tipo de complicaciones hemodinámicas son las que, lamentablemente, más ponen en riesgo la supervivencia del neonato.
Debido a que la enfermedad afecta a múltiples niveles —incluyendo el sistema renal, el gastrointestinal y el musculoesquelético—, el tratamiento no puede ser sencillo. Básicamente, se hace imprescindible contar con un enfoque multidisciplinario. Solo mediante la colaboración estrecha entre pediatras, genetistas, cardiólogos y especialistas en cuidados neonatales, se puede ofrecer un manejo clínico que trate de abordar de manera integral todas las necesidades de estos pacientes.
5. Diagnóstico
La detección del Síndrome de Edwards puede llevarse a cabo en momentos muy distintos, ya sea durante el embarazo o una vez que el bebé ha nacido. De hecho, el cribado prenatal se ha convertido en una herramienta fundamental para los especialistas. Básicamente, consiste en cruzar los datos obtenidos de los marcadores bioquímicos de la madre con los hallazgos ecográficos, lo que permite calcular con bastante precisión el riesgo de que existan alteraciones en los cromosomas del feto.
Si nos centramos en el primer trimestre, este proceso suele incluir una ecografía específica para medir la translucencia nucal. Por cierto, este dato no se analiza solo, sino que se combina con los niveles de ciertas sustancias en la sangre materna. Gracias a esto, es mucho más sencillo poner el foco en aquellos embarazos que presentan una mayor probabilidad de desarrollar esta patología.
La medicina, a través de la ciencia, no deja de avanzar. En los últimos años, han ganado muchísimo terreno las pruebas prenatales no invasivas, que se basan en el estudio del ADN fetal libre que circula por la sangre de la madre. A decir verdad, estas técnicas han supuesto un cambio radical, ya que ofrecen una sensibilidad y una especificidad altísimas a la hora de identificar la trisomía 18.
Por otro lado, si por cualquier motivo no se pudo realizar el seguimiento durante los primeros meses, algunos programas de salud pública ofrecen el llamado test cuádruple durante el segundo trimestre. Es una alternativa muy útil que permite mantener la vigilancia sobre la salud del bebé a medida que avanza la gestación.
A pesar de que las pruebas anteriores son de gran ayuda, para obtener una confirmación absoluta es necesario dar un paso más. De hecho, el diagnóstico definitivo solo se consigue mediante procedimientos de carácter invasivo, como la amniocentesis o la biopsia de vellosidades coriónicas.
Básicamente, lo que permiten estas intervenciones es tomar muestras para analizar directamente el cariotipo fetal mediante técnicas moleculares o citogenéticas. Al final, este análisis es el único que puede decirnos con total certeza qué está ocurriendo, permitiendo distinguir si nos encontramos ante una trisomía completa, una parcial o un cuadro de mosaico.
6. Pronóstico y esperanza de vida
Nos encontramos ante un panorama clínico bastante complejo y, por lo general, poco alentador para las familias. De hecho, la gran mayoría de los embarazos donde se detecta la trisomía 18 no llegan a completarse de forma natural, ya que suelen producirse abortos espontáneos o casos de muerte fetal intrauterina. Dicho de otro modo, el simple hecho de alcanzar el momento del parto ya representa el primer gran obstáculo biológico para estos fetos, debido a la fragilidad que impone la carga genética adicional.
Una vez que se produce el alumbramiento, la supervivencia de los recién nacidos suele ser, por desgracia, muy limitada. Por cierto, las estimaciones médicas actuales sitúan la esperanza de vida media en un margen que apenas oscila entre unos pocos días y unas cuantas semanas.
Es una realidad dura de asimilar: aproximadamente la mitad de los bebés fallecen durante su primera semana de vida. Sin embargo, no todo es una estadística cerrada; existe un pequeño porcentaje de pacientes que logra superar el primer año, aunque sigan siendo casos muy reducidos dentro del total de nacimientos.
No existe una regla fija para todos, ya que influyen diversos elementos en la evolución de cada pequeño. Por ejemplo, pesan mucho la gravedad de las malformaciones congénitas y, de manera muy especial, la presencia de cardiopatías complejas que comprometan el funcionamiento del corazón.
Otro factor clave es el tipo de alteración; de hecho, los casos identificados como trisomía en mosaico suelen presentar un camino algo distinto. Gracias a esto y al acceso a una atención médica especializada, se han llegado a documentar situaciones excepcionales donde la persona alcanza incluso la edad adulta, rompiendo con todas las expectativas habituales del síndrome.
En los últimos tiempos, el escenario ha empezado a cambiar ligeramente en ciertos centros de salud de vanguardia. Gracias a los constantes avances en cuidados neonatales y a una visión mucho más centrada en las necesidades de cada paciente, se han reportado mejoras en la permanencia de estos niños.
A pesar de este tratamiento individualizado, lo cierto es que la mortalidad sigue siendo extremadamente elevada. A decir verdad, el pronóstico continúa posicionándose como uno de los retos clínicos más significativos y difíciles de gestionar en la medicina contemporánea.
7. Tratamientos y cuidados médicos
Durante mucho tiempo, el enfoque médico que se aplicaba a quienes nacían con Síndrome de Edwards era fundamentalmente conservador. La lógica detrás de esto era la altísima tasa de mortalidad y la severidad de las malformaciones que presentaban los bebés. Sin embargo, lo cierto es que estamos siendo testigos de un cambio gradual en la manera de entender y abordar esta realidad. De hecho, ya no se ve todo de forma tan cerrada, y el manejo clínico está empezando a evolucionar hacia nuevas posibilidades.
El día a día en las unidades neonatales especializadas es, a decir verdad, sumamente complejo. Lo primero es garantizar la estabilidad del recién nacido, lo que suele implicar un esfuerzo constante en el soporte respiratorio y la estabilización cardiovascular, además de tratar cualquier complicación que surja sobre la marcha.
Por cierto, un obstáculo que suele aparecer muy pronto es la alimentación. Como estos bebés suelen tener serias dificultades de succión y deglución, lograr que reciban los nutrientes necesarios se convierte en una prioridad absoluta. Para conseguirlo, es habitual que los médicos tengan que recurrir a métodos como las sondas de alimentación o, en casos que requieren una solución a más largo plazo, las gastrostomías. Sin embargo, cada paso se da buscando siempre el bienestar del pequeño.
Uno de los temas que más debate genera hoy en día entre los especialistas es, sin duda, el papel de la cirugía. Especialmente cuando hablamos de las cardiopatías congénitas, la decisión de operar no es nada sencilla. De hecho, algunos estudios que han salido a la luz recientemente plantean que las intervenciones quirúrgicas selectivas podrían ser una opción viable, siempre y cuando se realice una evaluación muy rigurosa de cada paciente. No es una solución universal, pero abre una puerta que antes estaba prácticamente cerrada.
Pero no podemos olvidar que el manejo de este síndrome va mucho más allá de los quirófanos o los fármacos. El apoyo psicológico y el acompañamiento a las familias son, de hecho, piezas fundamentales en todo este proceso.
Al final, cualquier decisión terapéutica que se tome arrastra consigo consideraciones éticas de gran calado. Por eso, el enfoque actual intenta centrarse, por encima de todo, en la calidad de vida del paciente. Se trata de buscar un equilibrio humano en medio de una situación clínica tan delicada, donde la empatía cuenta tanto como el conocimiento médico.
8. Nuevas perspectivas médicas
La manera de entender el Síndrome de Edwards ha dado un vuelco impresionante en los últimos tiempos. De hecho, estamos dejando atrás esa idea de que solo se pueden ofrecer cuidados paliativos para pasar a un enfoque mucho más personal. A decir verdad, ya no se trata de aplicar una norma general a todo el mundo, sino de realizar una evaluación de cada caso de forma individualizada. Este cambio de mentalidad busca, por encima de todo, que las decisiones médicas se adapten a la realidad específica de cada paciente y no solo a un diagnóstico sobre el papel.
Por si fuera poco, el desarrollo de herramientas de diagnóstico ha avanzado una barbaridad. Hoy en día, gracias a la secuenciación genética y a los métodos que analizan el ácido desoxirribonucleico fetal libre en la sangre de la madre, es posible detectar la enfermedad con una precisión que antes era impensable. Sin embargo, lo más valioso de este progreso es que permite identificar la alteración en etapas muy tempranas del embarazo. Esto, lógicamente, les da a los especialistas y a los padres un margen de maniobra mucho mayor para entender a qué se enfrentan.
Estos saltos en la medicina neonatal han abierto puertas que antes estaban cerradas. Actualmente, se barajan opciones como las intervenciones quirúrgicas selectivas o el uso de soporte vital avanzado, algo que hace unos años ni siquiera se planteaba para estos niños. No obstante, esto también ha traído consigo una buena dosis de debate sobre hasta dónde debe llegar la intervención médica. Es un terreno complejo, pero lo positivo es que ahora existen más alternativas terapéuticas para intentar mejorar el pronóstico de algunos pacientes.
Una de las áreas de estudio que más interés está despertando últimamente es la que intenta descifrar el mosaicismo. Básicamente, los investigadores están tratando de comprender cómo influye el porcentaje de células trisómicas en la gravedad de los síntomas, lo que técnicamente llamamos el fenotipo clínico. De hecho, si logramos entender mejor esta relación, será mucho más fácil predecir el futuro de cada individuo de forma personalizada.
Al final, todos estos esfuerzos convergen en un mismo punto: mejorar la calidad de vida de quienes conviven con la enfermedad. Se trata de ofrecer a las familias una información más precisa para que, cuando llegue el momento de tomar decisiones clínicas importantes, sientan que tienen el control y el conocimiento necesario.
9. Conclusión
El Síndrome de Edwards se posiciona como una de las alteraciones cromosómicas más severas y difíciles de gestionar que conocemos. Básicamente, el problema surge cuando aparece una copia adicional del cromosoma 18.
Este material genético de más desencadena lo que podríamos llamar una reacción en cadena de errores en el desarrollo embrionario. Al final, esto se traduce en una multitud de malformaciones congénitas que terminan dañando varios sistemas orgánicos de forma simultánea. No es simplemente un fallo aislado, sino una alteración que afecta al organismo en su totalidad.
Es cierto que, en los últimos tiempos, hemos visto progresos increíbles tanto en las técnicas de diagnóstico prenatal como en los cuidados neonatales. Sin embargo, a pesar de estos logros tecnológicos, el pronóstico para estos pacientes sigue siendo, a decir verdad, bastante reservado.
De hecho, la elevada mortalidad que se registra tanto en la etapa fetal como en los primeros días de vida se mantiene como uno de los muros más difíciles de derribar para la comunidad médica. Por cierto, este sigue siendo hoy por hoy uno de los mayores desafíos científicos a los que nos enfrentamos en las unidades de neonatología.
Afortunadamente, no todo son malas noticias. El impulso constante de la investigación científica y el perfeccionamiento de las tecnologías diagnósticas nos han permitido comprender mucho mejor los mecanismos biológicos que rigen esta trisomía 18.
Lo bueno de este conocimiento es que nos está permitiendo dejar atrás las soluciones genéricas para centrarnos en estrategias clínicas más personalizadas. Además, esto ayuda enormemente a que el asesoramiento genético sea mucho más humano, cercano y preciso para las familias que se ven afectadas por este diagnóstico. Al final, tener información de calidad es la mejor herramienta para afrontar el camino.
Si miramos hacia adelante, está claro que la clave va a estar en saber combinar tres pilares fundamentales: la genética molecular, la medicina fetal y, por supuesto, la bioética. Esta enfermedad no solo nos obliga a buscar respuestas en el laboratorio, sino que nos invita a reflexionar profundamente sobre cómo se deben tomar las decisiones clínicas en situaciones tan delicadas.
En definitiva, el objetivo para los próximos años es consolidar un cuidado centrado en el paciente y su familia, donde el apoyo emocional y el respeto a la dignidad humana pesen tanto como los avances científicos.

10. Referencias
Benn, P., Borell, A., Chiu, R., et al. (2022). Position statement from the International Society for Prenatal Diagnosis on prenatal screening for trisomy 18. Prenatal Diagnosis, 42(11), 1361–1381. https://doi.org/10.1002/pd.6247
Benn, P., Cuckle, H., & Pergament, E. (2022). Non-invasive prenatal testing for aneuploidy: Current perspectives and future directions. Prenatal Diagnosis, 42(6), 724–731. https://doi.org/10.1002/pd.6152
Bianchi, D. W., Parker, R. L., et al. (2023). Noninvasive prenatal testing and detection of trisomy 18 in a large clinical cohort. Genetics in Medicine, 25(6), 100827. https://doi.org/10.1016/j.gim.2023.100827
Boghossian, N. S., et al. (2023). Epidemiology and survival of trisomy 18 in the United States, 2012–2021. American Journal of Medical Genetics Part A, 191(7), 1774–1783. https://doi.org/10.1002/ajmg.a.63185
Bruns, D. A., Campbell, E., & Wadhawan, R. (2022). Longitudinal developmental outcomes of children with trisomy 18: A 5-year follow-up. Journal of Pediatric Genetics, 11(04), 282–289. https://doi.org/10.1055/s-0040-1721074
Bruns, D. A., Campbell, E., & Wadhawan, R. (2023). Health outcomes and service use of children with trisomy 18. American Journal of Medical Genetics Part A, 191(1), 107–114. https://doi.org/10.1002/ajmg.a.62985
Bruns, D. A., et al. (2024). Quality of life in children with trisomy 18: Parent perspectives and longitudinal data. Journal of Pediatric Nursing, 75, 54–61. https://doi.org/10.1016/j.pedn.2023.12.016
Carey, J. C. (2022). Trisomy 18 and trisomy 13 syndromes: History, phenotype, and management. Clinics in Perinatology, 49(2), 315–331. https://doi.org/10.1016/j.clp.2022.02.002
Choi, H., et al. (2024). Genetic characterization of trisomy 18 using next-generation sequencing: A comprehensive analysis. Genes, 15(1), 89. https://doi.org/10.3390/genes15010089
Costello, J. M., et al. (2023). Surgical outcomes in congenital heart disease associated with trisomy 18: A multicenter study. The Journal of Thoracic and Cardiovascular Surgery, 166(5), 1431–1442. https://doi.org/10.1016/j.jtcvs.2023.01.031
Dowaikh, A., Alsahari, A., Khoshhal, S., & Momenah, T. (2024). Cardiac care in trisomy 18: A path to improved outcomes. Journal of Taibah University Medical Sciences, 19(3), 545–548. https://doi.org/10.1016/j.jtumed.2024.04.003
Fujimoto, A., et al. (2022). Long-term survivors with trisomy 18: Clinical characteristics and survival analysis of 50 cases. American Journal of Medical Genetics Part A, 188(12), 3465–3473. https://doi.org/10.1002/ajmg.a.62969
Ghi, T., et al. (2022). Ultrasound markers of trisomy 18 in the first trimester: A systematic review. Ultrasound in Obstetrics & Gynecology, 60(5), 624–631. https://doi.org/10.1002/uog.26048
Gil, M. M., Quezada, M. S., Revello, R., Akolekar, R., & Nicolaides, K. H. (2022). Analysis of cell-free DNA testing for trisomy 18, 13, and 21. Ultrasound in Obstetrics & Gynecology, 59(1), 10–15. https://doi.org/10.1002/uog.24823
Graham, E. M., Bradley, S. M., et al. (2022). Outcomes after cardiac surgery in trisomy 18: A 10-year experience. Pediatric Cardiology, 43, 1058–1066. https://doi.org/10.1007/s00246-022-02826-6
Guon, J., Wilfond, B. S., Farlow, B., Brazg, T., & Janvier, A. (2022). Our children are not a diagnosis: Ethical perspectives on trisomy 18 care. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 190(3), 263–270. https://doi.org/10.1002/ajmg.c.31985
Hashimoto, Y., et al. (2022). Ethical considerations in the treatment of trisomy 18: A Japanese perspective. Bioethics, 36(6), 661–669. https://doi.org/10.1111/bioe.13014
Imataka, G., et al. (2022). Clinical characteristics of mosaic trisomy 18: Case report and review. Clinical Case Reports, 10(4), e05668. https://doi.org/10.1002/ccr3.5668
Janvier, A., et al. (2022). Parental perspectives on trisomy 18 care: A global survey. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 190(3), 329–338. https://doi.org/10.1002/ajmg.c.31993
Janvier, A., Farlow, B., & Wilfond, B. S. (2023). The experience of families raising children with trisomy 18. Pediatrics, 151(1), e2022058567. https://doi.org/10.1542/peds.2022-058567
Kaneko, Y., Kobayashi, J., et al. (2023). Outcomes of surgical treatment in trisomy 18 infants with congenital heart disease. European Journal of Cardio-Thoracic Surgery, 64(4), ezad325. https://doi.org/10.1093/ejcts/ezad325
Kato, E., et al. (2022). Medical interventions in trisomy 18 infants: Survival and outcomes. Pediatrics International, 64(1), e15102. https://doi.org/10.1111/ped.15102
Khalil, A., et al. (2022). Fetal growth restriction and trisomy 18: Risk and management. Ultrasound in Obstetrics & Gynecology, 60(2), 200–211. https://doi.org/10.1002/uog.24941
Kosho, T., Nakamura, T., Kawame, H., Baba, A., Tamura, M., & Fukushima, Y. (2022). Natural history of trisomy 18: Survival, clinical manifestations and management. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 190(3), 295–311. https://doi.org/10.1002/ajmg.c.31998
Kosho, T., et al. (2023). Clinical management guidelines for trisomy 18: 2023 update. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 193(3), e32057. https://doi.org/10.1002/ajmg.c.32057
Li, Y., et al. (2022). Genomic insights into trisomy 18: A molecular study. Human Genomics, 16, 17. https://doi.org/10.1186/s40246-022-00392-4
McDowell, M., et al. (2024). Neonatal intensive care in trisomy 18: A retrospective study. Journal of Neonatal-Perinatal Medicine, 17(2), 215–224. https://doi.org/10.3233/NPM-230132
McGraw, M. P., et al. (2024). Advances in prenatal screening for trisomy 18 using cell-free DNA testing. Prenatal Diagnosis, 44(1), 31–40. https://doi.org/10.1002/pd.6482
Merritt, T. A., et al. (2022). Neonatal management strategies for trisomy 18: Consensus and controversies. Neonatology, 119(4), 433–443. https://doi.org/10.1159/000524588
Meyer, R. E., et al. (2022). Population trends in trisomy 18 survival: A multi-state study. Birth Defects Research, 114(15), 903–912. https://doi.org/10.1002/bdr2.2045
Meyer, R. E., Liu, G., et al. (2023). Population survival estimates for trisomy 18 in the modern era. Birth Defects Research, 115(16), 1461–1471. https://doi.org/10.1002/bdr2.2231
Meyer, R. E., Liu, G., Gilboa, S. M., et al. (2023). Survival of infants with trisomy 18 in population-based birth defect surveillance programs. Birth Defects Research, 115(2), 209–221. https://doi.org/10.1002/bdr2.2120
Nakano, Y., et al. (2023). Outcomes of aggressive treatment strategies in trisomy 18. Journal of Perinatology, 43, 903–910. https://doi.org/10.1038/s41372-023-01646-0
Nelson, K. E., et al. (2022). Palliative versus intensive care in trisomy 18: Parental decision-making. The Journal of Pediatrics, 247, 51–58. https://doi.org/10.1016/j.jpeds.2022.04.032
Nelson, K. E., Rosella, L. C., Mahant, S., & Guttmann, A. (2022). Survival and surgical interventions for children with trisomy 18. Pediatrics, 150(1), e2021055866. https://doi.org/10.1542/peds.2021-055866
Nelson, K. E., Hexem, K. R., & Feudtner, C. (2023). Invasive interventions in children with trisomy 18: A national study. The Journal of Pediatrics, 259, 113426. https://doi.org/10.1016/j.jpeds.2023.113426
Norton, M. E., Biggio, J. R., et al. (2022). Cell-free DNA analysis for noninvasive prenatal screening: Updated clinical guidelines. New England Journal of Medicine, 386, 1619–1631.
Paladini, D., et al. (2022). Fetal cardiac anomalies associated with trisomy 18: A detailed analysis. Prenatal Diagnosis, 42(11), 1445–1454. https://doi.org/10.1002/pd.6242
Peterson, J. K., Kochilas, L., Catton, K. G., Moller, J. H., & Setty, S. P. (2022). Long-term outcomes of children with trisomy 18 receiving medical interventions. Congenital Heart Disease, 17(2), 161–171. https://doi.org/10.32604/chd.2022.18182
Peterson, J. K., et al. (2023). Cardiac surgery outcomes in trisomy 18 patients: A retrospective review. The Annals of Thoracic Surgery, 116(1), 108–115. https://doi.org/10.1016/j.athoracsur.2022.09.020
Puri, K., et al. (2023). Clinical spectrum of trisomy 18 in neonates: A multicenter study. BMC Pediatrics, 23, 237. https://doi.org/10.1186/s12887-023-04061-4
Rasmussen, S. A., Wong, L. Y., Yang, Q., May, K. M., & Friedman, J. M. (2022). Population-based analyses of trisomy 18 outcomes. Genetics in Medicine, 24(1), 127–134. https://doi.org/10.1016/j.gim.2021.08.012
Sago, H., et al. (2022). Prenatal ultrasound findings in trisomy 18: A Japanese study. Journal of Obstetrics and Gynaecology Research, 48(4), 896–903. https://doi.org/10.1111/jog.15174
Saito, K., et al. (2023). Advances in prenatal diagnosis of trisomy 18: A comprehensive review. Prenatal Diagnosis, 43(1), 5–15. https://doi.org/10.1002/pd.6288
Schlosser, M. P., MacPherson, M. J., Castro-Codesal, M., Mack, C., Sue-Milne, K., Wren, T., Shapka, L., Kung, J. Y., & van Manen, M. (2024). Liveborn children with trisomy 18: A scoping review. Journal of Neonatal-Perinatal Medicine, 17(1), 5–22. https://doi.org/10.1177/19345798241302276
Sepulveda, W., et al. (2023). Fetal anomalies detected in trisomy 18 pregnancies: A 20-year experience. Prenatal Diagnosis, 43(4), 481–490. https://doi.org/10.1002/pd.6310
Shravya, M. S., Girisha, K. M., & Nayak, S. S. (2024). Comprehensive phenotyping of fetuses with trisomy 18: A perinatal center experience. Clinical Dysmorphology, 33(1), 16–26. https://doi.org/10.1097/MCD.0000000000000481
Sun, W., Wang, L., Liu, T., Ouyang, L., Qin, W., Luo, J., & Qin, Z. (2023). Phenotypic spectrum of trisomy 18 mosaicism: A new patient and literature review. Clinical Laboratory, 69(3). https://doi.org/10.7754/Clin.Lab.2022.220610
Wang, X., et al. (2023). Molecular cytogenetic analysis of trisomy 18 cases: A study of 120 cases. Molecular Cytogenetics, 16, 21. https://doi.org/10.1186/s13039-023-00650-6
Wang, Y., Chen, Y., Tian, F., et al. (2023). Maternal plasma DNA sequencing in prenatal diagnosis of trisomy 18. Scientific Reports, 13, 8932. https://doi.org/10.1038/s41598-023-35921-2
Wapner, R. J., Martin, C. L., et al. (2022). Chromosomal microarray versus karyotyping in prenatal diagnosis: Current practice. New England Journal of Medicine, 386, 1215–1224.
Wilkinson, D., & de Crespigny, L. (2023). Ethical debates around treatment of trisomy 18: A systematic approach. Journal of Medical Ethics, 49(12), 823–824. https://doi.org/10.1136/jme-2023-109312
Yates, R., et al. (2023). Congenital heart disease in trisomy 18: A cohort study of 150 patients. Cardiology in the Young, 33(7), 1210–1216. https://doi.org/10.1017/S104795112200251X
Zhang, H., Gao, Y., Jiang, F., et al. (2023). Non-invasive prenatal testing for trisomy 18 in large cohorts: A multicenter study. Molecular Cytogenetics, 16, 5. https://doi.org/10.1186/s13039-023-00635-5
Ziogas, I. A., Kakos, C. D., Kokkinakis, S., Hills-Dunlap, J. L., Corkum, K. S., Acker, S. N., Diaz-Miron, J. L., Lovvorn, H. N., Roach, J. P., & Gosain, A. (2024). Management and outcomes of hepatoblastoma in patients with trisomy 18: A systematic review and pooled analysis. Journal of Pediatric Surgery, 59(4), 661–667. https://doi.org/10.1016/j.jpedsurg.2023.10.027