


Síndrome de West: causas, síntomas y por qué el diagnóstico precoz lo cambia todo
José Hernández Jiménez
4/9/202616 min leer


1. Introducción
El síndrome de West, conocido también como el síndrome de espasmos infantiles, representa una de las encefalopatías epilépticas más serias que pueden afectar a un bebé. De hecho, esta enfermedad suele dar sus primeras señales de forma muy precoz, apareciendo generalmente durante el primer año de vida.
A decir verdad, su importancia en el ámbito médico ha crecido enormemente en los últimos tiempos. Esto se debe, en gran medida, al gran avance de las técnicas diagnósticas genéticas y al desarrollo de nuevas estrategias terapéuticas dirigidas, que permiten abordar el problema de una manera mucho más precisa que hace unos años.
En la práctica clínica diaria existe un obstáculo importante: la dificultad de identificar los síntomas correctamente. Es muy común, por desgracia, que se confundan los espasmos epilépticos con movimientos naturales del lactante, como los sobresaltos normales o el conocido reflejo de Moro.
Esta confusión no es un tema menor, ya que suele provocar un retraso del diagnóstico que puede ser fatal. Por cierto, la evidencia médica actual es muy clara al respecto: el tiempo hasta el inicio del tratamiento es, probablemente, el factor más determinante para el pronóstico y el futuro del pequeño.
Desde una perspectiva médica, no hay duda de que este síndrome debe tratarse como una auténtica urgencia neurológica. El problema principal radica en la presencia de una actividad epileptiforme desorganizada, un fenómeno técnico denominado hipsarritmia.
Esta anomalía eléctrica es tan persistente que termina interfiriendo directamente con la maduración cerebral. Si no se interviene a tiempo, el resultado suele ser una regresión o detención del desarrollo psicomotor. En definitiva, el cerebro del lactante se ve sometido a un caos eléctrico que impide que sus capacidades avancen como deberían, lo que subraya la necesidad de actuar con la máxima rapidez posible.
2. ¿Qué es el síndrome de West?
Para empezar, debemos entender que el síndrome de West no es una afección común; se trata de una encefalopatía epiléptica que da la cara muy pronto en la vida del bebé. Lo que define a este cuadro es lo que los especialistas llaman una tríada clásica. Esta combinación de síntomas incluye los espasmos epilépticos, un patrón de actividad cerebral muy desorganizado conocido como hipsarritmia —que se detecta mediante un electroencefalograma— y, lamentablemente, un deterioro o regresión del desarrollo neurológico.
¿Cómo se manifiestan estos episodios en el día a día? Lo cierto es que los ataques suelen presentarse como contracciones breves y simétricas que afectan al cuello, el tronco y las extremidades del pequeño. De hecho, es muy habitual que estos movimientos no ocurran de forma aislada, sino en salvas o agrupamientos de varios espasmos seguidos.
Sin embargo, hay un detalle que suele complicar las cosas para los médicos y las familias: no siempre aparecen los tres síntomas de la tríada al mismo tiempo cuando la enfermedad está empezando. Por cierto, esta falta de sincronía es, a menudo, lo que dificulta un diagnóstico temprano, que es vital para el futuro del paciente.
Dicho esto, la forma de entender este problema ha evolucionado mucho últimamente. Hoy en día, se reconoce que no estamos ante una enfermedad única con una sola causa, sino ante un fenotipo común. Básicamente, lo que ocurre es que existen múltiples etiologías o causas diferentes que acaban provocando un fallo similar en las redes neuronales inmaduras del lactante.
Esta perspectiva ha transformado el modo de clasificar el síndrome, apostando por un enfoque mecanístico que intenta comprender el "cómo" y el "por qué" ocurre la falla, más allá de simplemente describir los síntomas. De hecho, se ha propuesto utilizar el término síndrome de espasmos epilépticos infantiles para reflejar mejor esta heterogeneidad en sus causas y su naturaleza compleja. Al final, entender que es un síndrome con diversos orígenes es el primer paso para un abordaje mucho más preciso.
3. Cómo reconocer los síntomas
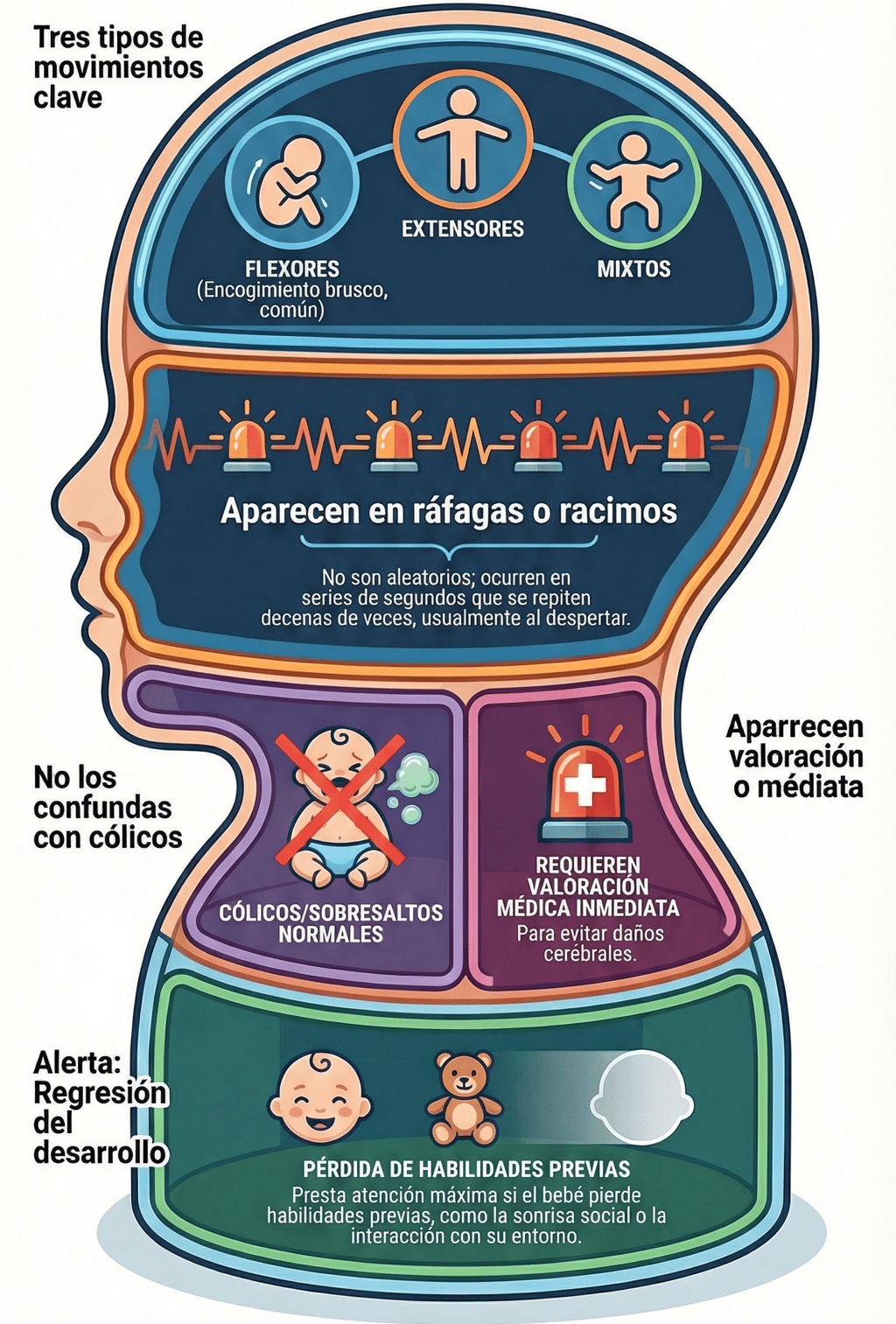
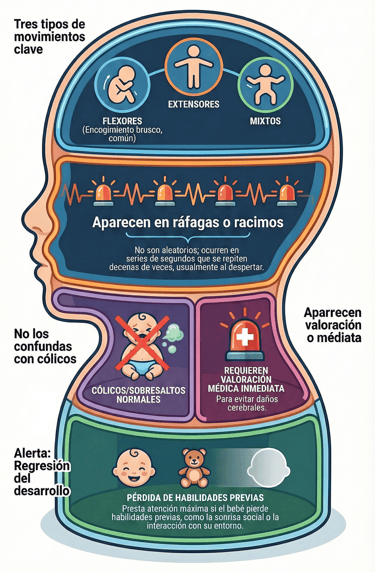
imagen: propia
A decir verdad, identificar los espasmos infantiles puede ser todo un reto al principio porque sus señales suelen ser bastante sutiles. Sin embargo, la medicina los clasifica principalmente en tres tipos: flexores, extensores y mixtos.
De hecho, los que se ven con más frecuencia son los de tipo flexor. Estos se reconocen por un encogimiento brusco del tronco acompañado de una flexión de las extremidades. Aunque parezcan movimientos aislados, entender esta clasificación es el primer paso para saber ante qué nos encontramos.
Un rasgo que resulta fundamental para dar la voz de alarma es la forma en que se presentan. Por cierto, estos movimientos no suelen ocurrir de manera aleatoria, sino que aparecen en racimos o grupos.
Es muy común que estos episodios sucedan especialmente al despertar o justo tras el sueño del bebé. Aunque cada espasmo individual apenas dura unos pocos segundos, lo que realmente constituye una señal de alarma clínica es que pueden llegar a repetirse decenas de veces en una misma serie. Ver al pequeño encadenar tantos movimientos seguidos es, sin duda, un motivo de peso para buscar valoración médica inmediata.
A diferencia de lo que ocurre con las convulsiones más clásicas y llamativas, los espasmos tienen una trampa: se pueden interpretar erróneamente con mucha facilidad. Es muy habitual que los padres o cuidadores los confundan con simples cólicos o incluso con movimientos normales del lactante.
Desgraciadamente, esta confusión suele provocar un retraso diagnóstico, lo que se convierte en uno de los factores que más influyen en un peor pronóstico. Por eso, ante la duda, siempre es mejor pecar de precavidos que dejar pasar el tiempo.
Finalmente, hay otro signo de gran relevancia que va más allá de lo físico: la regresión del desarrollo. De hecho, hay que prestar muchísima atención si el bebé empieza a sufrir una pérdida de habilidades que ya había adquirido previamente.
Esto se nota claramente cuando hay una disminución de la interacción social o algo tan característico como la pérdida de la sonrisa social. Estos cambios en su forma de relacionarse con el entorno son indicadores clave de que algo en su sistema neurológico necesita atención urgente.
4. Qué ocurre en el cerebro
Si hay algo que define visualmente al síndrome de West en las pruebas médicas, es la hipsarritmia. Este es el hallazgo principal que los especialistas buscan en un electroencefalograma. De hecho, se trata de un patrón caótico y completamente desorganizado, con una alta amplitud que no es más que el reflejo de una actividad neuronal anormal que afecta al cerebro de manera generalizada. Es, básicamente, una tormenta eléctrica que impide el funcionamiento normal.
¿Por qué se produce este fenómeno? Lo cierto es que, desde un punto de vista fisiopatológico, se ha demostrado que existen múltiples causas que pueden desencadenarlo. Ya hablemos de factores de origen genético, estructural o incluso metabólico, todos ellos terminan coincidiendo en un mismo problema: una disfunción de las redes neuronales. Esta falla de comunicación involucra zonas críticas para el desarrollo, como la corteza, el tálamo y el tronco encefálico. Al final, diferentes caminos llevan a un mismo error en el sistema.
Por cierto, hay un componente químico muy relevante detrás de este cuadro. Se ha observado que existe un marcado desequilibrio entre los neurotransmisores excitatorios e inhibitorios. Sin embargo, la cosa no queda ahí, ya que también se han detectado alteraciones en factores neurotróficos esenciales, concretamente en el BDNF y en el IGF-1.
Este entorno tan alterado es, en última instancia, el que favorece la hiperexcitabilidad neuronal. Es precisamente este estado de agitación constante lo que da pie a la aparición de los espasmos.
Lo más delicado de este asunto es que esta actividad epileptiforme persistente no es un evento aislado, sino que tiene consecuencias a largo plazo. Al ser tan constante, termina interfiriendo directamente con procesos vitales como la sinaptogénesis —la creación de nuevas conexiones— y la plasticidad cerebral.
Esto explica, de hecho, por qué el síndrome tiene ese impacto negativo en el desarrollo cognitivo. Básicamente, el cerebro está tan ocupado lidiando con el caos eléctrico que no puede dedicarse a construir las redes necesarias para el aprendizaje y el crecimiento normal.
5. Causas del síndrome
Las causas del síndrome de West son altamente heterogéneas y se agrupan en tres categorías principales:
Genéticas
Para entender realmente qué está pasando en el organismo, tenemos que echar un vistazo a lo que ocurre en lo más profundo de nuestras células. De hecho, hoy sabemos que las mutaciones en puntos muy específicos de nuestro ADN son las responsables directas de este cuadro clínico. Por cierto, entre los nombres que más suenan en los estudios actuales aparecen genes como el STXBP1, el KCNQ2 o el conjunto de los GRIN2A/B.
Sin embargo, la genética es compleja y no se limita a un par de nombres aislados. También se ha comprobado que todos aquellos genes que están vinculados de una forma u otra con el complejo de esclerosis tuberosa actúan como causas relevantes que no podemos pasar por alto. Básicamente, estos hallazgos permiten a los especialistas ponerle nombre y apellido al origen del problema, algo fundamental para comprender la raíz de la enfermedad.
Lesiones cerebrales
Cuando analizamos por qué se producen estos casos, nos encontramos con un abanico de causas muy específicas que aparecen de forma recurrente. Por cierto, estas situaciones representan una proporción significativa de los diagnósticos globales, por lo que entender su origen es fundamental para cualquier abordaje médico.
Entre los factores más relevantes, destacan sin duda las malformaciones corticales, que afectan a la estructura misma del cerebro. Sin embargo, el panorama es amplio. También juegan un papel crucial las secuelas derivadas de una encefalopatía hipóxico-isquémica, un evento que marca un antes y un después en la salud del paciente. Además, no podemos pasar por alto el impacto que pueden tener las infecciones o los traumatismos físicos accidentales, ya que ambos son elementos que explican gran parte de la casuística actual.
Idiopáticas
Sin duda, dar con la raíz del problema es un paso fundamental en cualquier proceso médico. De hecho, la identificación etiológica es la verdadera brújula que guía a los especialistas para decidir qué selección terapéutica es la más acertada para cada caso. No es solo una cuestión de poner un nombre a lo que ocurre; sin embargo, entender el origen ayuda enormemente a definir el pronóstico y saber qué esperar en el futuro.
Por cierto, hay situaciones en las que, a pesar de realizar todas las pruebas necesarias, no se logra dar con una causa clara. Aunque pueda parecer preocupante, lo cierto es que estos casos suelen tener un mejor pronóstico.
Esto sucede especialmente cuando el organismo muestra una buena respuesta al tratamiento inicial. Al final, aunque el motivo original sea un misterio, que la medicina logre controlar los síntomas de forma eficaz es lo que realmente marca la diferencia en la evolución del paciente.
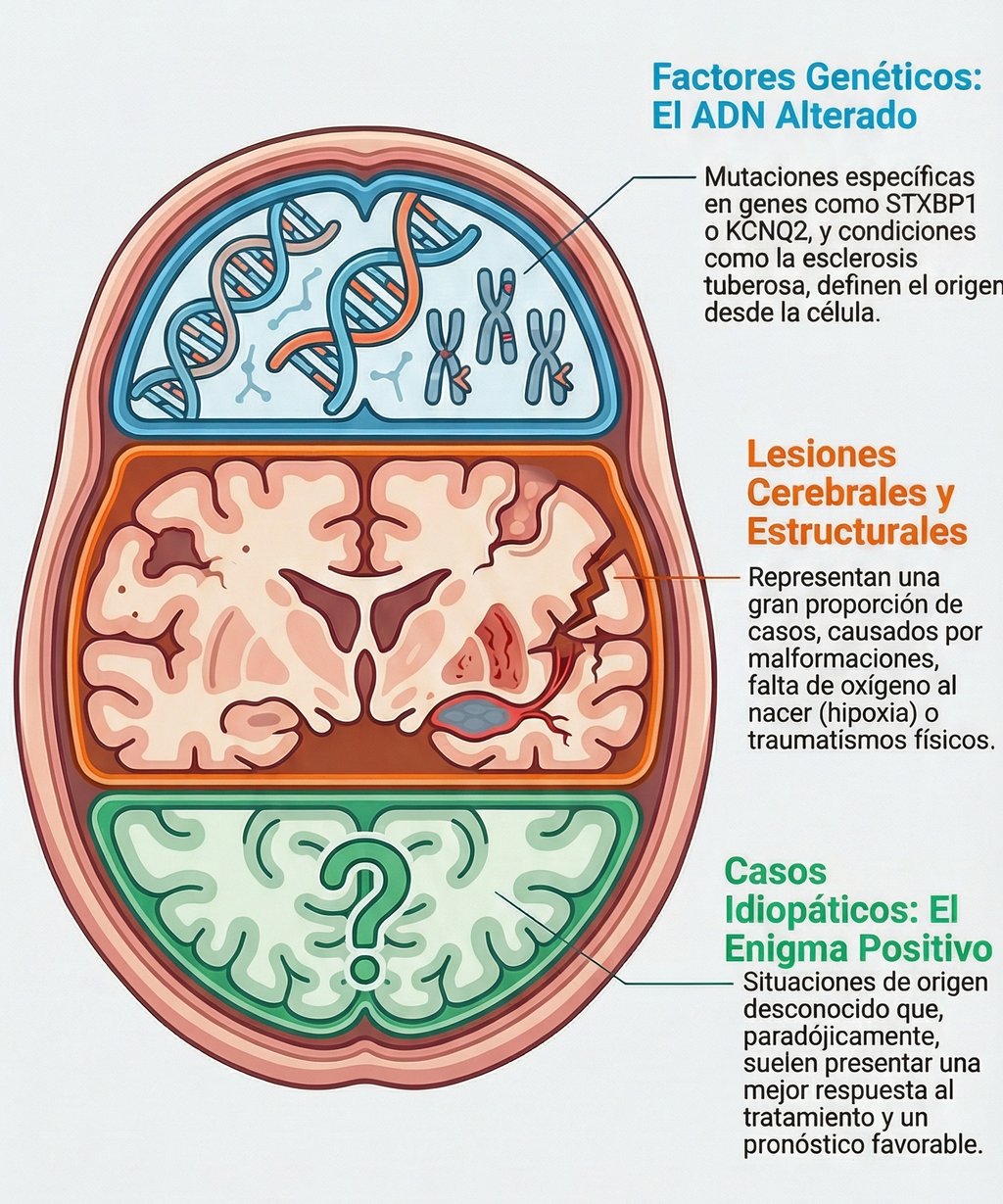
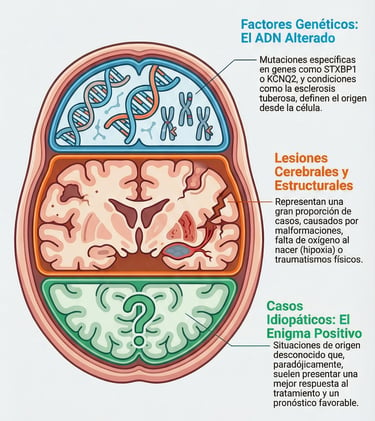
imagen: propia
6. Diagnóstico
Para confirmar la presencia del síndrome de West los especialistas no se apoyan en una sola señal, sino que cruzan lo que observan en el paciente con diversas pruebas complementarias. De hecho, este rompecabezas diagnóstico combina la observación de los síntomas con herramientas tecnológicas avanzadas.
El electroencefalograma resulta una pieza fundamental en todo este proceso; sin esta prueba sería prácticamente imposible identificar con precisión la hipsarritmia, que es esa actividad cerebral tan desorganizada y típica de este cuadro.
Para entender si existen causas estructurales que estén originando el problema, se recurre a la neuroimagen, empleando habitualmente la resonancia magnética.
Por otro lado, es fascinante ver cómo los estudios genéticos han ido ganando terreno hasta volverse piezas clave en el enfoque médico moderno. Gracias a ellos, hoy es posible profundizar en el origen biológico de la enfermedad con una precisión que antes era impensable.
Si hay algo en lo que todos los expertos coinciden, es en que el tiempo es un factor determinante. El diagnóstico precoz no es solo una recomendación, sino algo totalmente esencial para el futuro del niño.
Sin duda, la evidencia médica es muy clara al respecto: cuanto más temprano sea el inicio del tratamiento, mejores y más significativos serán los resultados finales. Al final, detectar el problema a tiempo permite que las intervenciones funcionen mucho mejor, marcando una diferencia real en la evolución del pequeño.
7. Tratamiento
El tratamiento del síndrome de West ha evolucionado, aunque persisten varias opciones principales:
Fármacos principales
El funcionamiento de estas intervenciones se basa en su capacidad para regular la excitabilidad neuronal. Para conseguirlo, los fármacos trabajan modulando directamente los neurotransmisores o influyendo en los diversos ejes hormonales que participan en la actividad del cerebro. Básicamente, se trata de ajustar estos procesos químicos para recuperar el equilibrio en el sistema nervioso.
Si nos detenemos a analizar lo que dicen los estudios comparativos, encontramos datos muy interesantes sobre cuál es el mejor camino a seguir. De hecho, la evidencia actual sugiere que las terapias hormonales pueden resultar más eficaces, al menos si miramos los resultados a corto plazo, en comparación con el uso de la vigabatrina para ciertos casos. Sin embargo, como siempre ocurre en medicina, la elección final dependerá de las circunstancias particulares de cada paciente.
Importancia del tiempo
Cuando nos enfrentamos a esta situación, la rapidez es nuestra mejor aliada. De hecho, el inicio precoz del tratamiento se ha consolidado como uno de los factores más determinantes para el pronóstico final del niño. No es una exageración; sin embargo, a veces se subestima la urgencia real de la intervención.
Por cierto, debemos ser muy conscientes de que el margen de maniobra es sumamente estrecho. Los retrasos en la atención, incluso si hablamos de apenas unas pocas semanas, tienen el potencial de impactar negativamente en el desarrollo general del pequeño. Al final, cada día que se gana es una oportunidad vital para proteger su futuro neurológico
Terapias complementarias
Existen medidas complementarias a los fármacos actuales. De hecho, una de las opciones que suele ponerse sobre la mesa es la dieta cetogénica, un régimen alimenticio muy específico que ayuda a controlar la actividad cerebral. Sin embargo, no es la única vía; en situaciones muy concretas y tras una evaluación exhaustiva, la cirugía puede ser una solución definitiva para esos casos seleccionados donde el origen está muy localizado.
Por otro lado, la medicina actual está avanzando con fuerza hacia lo que conocemos como terapias dirigidas. La idea es sencilla pero potente: aplicar soluciones pensadas específicamente para etiologías específicas.
Un ejemplo claro de este avance es el uso de los inhibidores mTOR. Estos compuestos se utilizan de forma muy dirigida cuando se detecta que el problema proviene del TSC. Al final, se trata de dejar de usar un enfoque general para pasar a una medicina mucho más personalizada y eficaz.
8. Pronóstico
Cuando hablamos del pronóstico del síndrome de West no es algo escrito en piedra; es sumamente variable y depende de varios factores que inclinan la balanza. De hecho, uno de los puntos más importantes es la etiología, es decir, el origen de la afección. Por cierto, los casos que se clasifican como idiopáticos —aquellos donde no se encuentra una causa subyacente clara— suelen tener una evolución bastante más favorable que el resto.
Sin embargo, no todo depende del origen. Otros dos pilares fundamentales que dictan cómo avanzará el paciente son el tiempo transcurrido hasta el tratamiento y, por supuesto, la capacidad de la persona para mostrar una buena respuesta terapéutica una vez iniciada la medicación.
A pesar de los avances, hay que ser realistas: una proporción significativa de los pacientes suele enfrentar diversas secuelas neurológicas a lo largo de su vida. No es un camino sencillo, ya que estas complicaciones pueden manifestarse de distintas maneras.
Es muy común observar, por ejemplo, la aparición de una discapacidad intelectual o el desarrollo de una epilepsia refractaria, que es aquella que resulta especialmente difícil de controlar con fármacos. Además, también es frecuente que estos niños presenten trastornos del espectro autista, lo que añade una capa extra de complejidad al cuidado y seguimiento que requieren.
Pero no todo es negativo. Existe un dato realmente esperanzador que debemos tener siempre presente: la rapidez con la que actuamos cambia radicalmente las reglas del juego.
De hecho, los casos que logran un diagnóstico y tratamiento precoz tienen una oportunidad de oro. La evidencia nos dice que, cuando se interviene a tiempo, estos pacientes pueden llegar a alcanzar un desarrollo cercano a la normalidad. Al final, la diferencia entre un futuro con graves limitaciones y una vida plena suele estar en la velocidad con la que los especialistas logran identificar y abordar el problema.
9. Nuevas investigaciones
El rumbo que está tomando la ciencia en los últimos tiempos es realmente prometedor. La tendencia actual se inclina claramente hacia lo que conocemos como medicina de precisión. Básicamente, este enfoque consiste en que el tratamiento deje de ser una fórmula general y pase a adaptarse de forma minuciosa a la causa subyacente de cada paciente. Es, por así decirlo, un traje a medida para cada caso.
Las investigaciones más recientes están poniendo toda su atención en las terapias dirigidas que tienen su base en la genética. Un ejemplo muy claro de este avance es el uso de fármacos como el everolimus. Este medicamento actúa como uno de los inhibidores de la diana de rapamicina en células de mamífero y resulta especialmente relevante cuando nos encontramos ante el complejo de esclerosis tuberosa.
Sin embargo, el campo de estudio es todavía más amplio. También se está trabajando intensamente en la modulación de los canales iónicos, centrándose en elementos específicos como el KCNQ o los receptores N-metil-D-aspartato. Todo este esfuerzo busca entender y corregir los fallos eléctricos del cerebro desde la raíz misma de la célula.
No podemos olvidar el papel fundamental que están jugando las nuevas herramientas tecnológicas. El uso de biomarcadores, junto con las técnicas de neuroimagen avanzada, está permitiendo que los especialistas realicen un diagnóstico mucho más fino.
De hecho, estas tecnologías no solo sirven para detectar el problema al principio, sino que son piezas clave para realizar un seguimiento constante y ver cómo evoluciona el paciente en el tiempo. Al final, contar con estas imágenes de alta resolución y con indicadores biológicos precisos es lo que está permitiendo mejorar significativamente la calidad de vida de las personas afectadas.


imagen: propia
10. Conclusión
El síndrome de West destaca sin duda como una de las encefalopatías epilépticas más críticas de la infancia. La preocupación no es para menos; de hecho, su importancia radica tanto en el profundo impacto que tiene sobre el desarrollo del niño como en la urgencia absoluta que requiere su abordaje médico. No es algo que pueda esperar, ya que cada momento cuenta.
Hoy por hoy, la evidencia científica es muy clara: el factor que más influye para mejorar el pronóstico de estos pequeños es, sin discusión, la detección y el tratamiento precoz. No basta con actuar, hay que hacerlo rápido.
Por cierto, el modo en que los médicos enfrentan esta enfermedad está dando un giro radical gracias a la identificación etiológica. En este aspecto, los estudios genéticos están siendo fundamentales. Gracias a ellos, el enfoque terapéutico está dejando de ser generalista para transformarse en una serie de estrategias personalizadas que van directas a la raíz del problema.
Sin embargo, hay que ser realistas y reconocer que, a pesar de los grandes pasos que se han dado, todavía persisten desafíos considerables. Uno de los mayores problemas es la enorme variabilidad en la respuesta al tratamiento; básicamente, lo que funciona de maravilla en un paciente puede no tener el mismo efecto en otro. Además, no podemos olvidar que, lamentablemente, en muchos casos todavía nos enfrentamos a una falta de terapias curativas definitivas, lo que obliga a seguir investigando sin descanso.
¿Hacia dónde nos dirigimos entonces? Lo cierto es que el futuro del manejo de esta condición apunta directamente hacia la medicina de precisión. De hecho, este nuevo paradigma se apoya en dos pilares que son innegociables: el diagnóstico temprano y el uso de terapias dirigidas. El objetivo final es que cada niño reciba un tratamiento diseñado específicamente para su caso, maximizando así sus oportunidades de recuperación y bienestar.

11. Referencias
Curatolo, P., Moavero, R., & de Vries, P. J. (2022). mTOR inhibitors in tuberous sclerosis. The Lancet Neurology, 21(5), 450–463.
Curatolo, P., Moavero, R., & Specchio, N. (2023). Advances in treatment of developmental epileptic encephalopathies. Current Opinion in Neurology, 36(2), 101–108.
Dulac, O., Nabbout, R., & Plouin, P. (2022). Epileptic encephalopathy in children. Handbook of Clinical Neurology, 188, 211–225.
Helbig, I., Heinzen, E. L., & Mefford, H. C. (2023). Genetic landscape of epileptic encephalopathies. Nature Genetics, 55(2), 200–210.
Hollenshead, P. P., Jackson, C. N., Cross, J. V., et al. (2023). Treatment modalities for infantile spasms: Current considerations and evolving strategies. Neurological Sciences, 44(11), 3823–3836.
Howell, K. B., Eggers, S., Dalziel, K., et al. (2023). A population-based cost-effectiveness study of early genetic testing in epilepsy. Neurology, 100(21), e2155–e2167.
Howell, K. B., et al. (2023). Genetic testing in epileptic encephalopathies. Neurology, 101(4), 160–172.
Kato, M. (2022). Molecular genetics of infantile epileptic encephalopathies. Brain and Development, 44(1), 2–11.
Knupp, K. G., Coryell, J., Nickels, K. C., et al. (2022). Response to treatment in infantile spasms. Neurology, 98(24), e2429–e2440.
Lux, A. L., Edwards, S. W., Hancock, E., et al. (2022). The United Kingdom Infantile Spasms Study (UKISS). The Lancet Neurology, 21(1), 15–16.
Nabbout, R., & Kuchenbuch, M. (2023). Precision medicine in epilepsy. Nature Reviews Neurology, 19(3), 150–163.
O'Callaghan, F. J. K., Edwards, S. W., Alber, F. D., et al. (2022). The aetiology of infantile spasms and its relevance to treatment. Epilepsia, 63(11), 2822–2834.
Osborne, J. P., Lux, A. L., & Edwards, S. W. (2022). The clinical spectrum of infantile spasms. Developmental Medicine & Child Neurology, 64(2), 160–166.
Pavone, P., Polizzi, A., Marino, S. D., Corsello, G., Falsaperla, R., Marino, S., & Ruggieri, M. (2020). West syndrome: A comprehensive review. Neurological Sciences, 41(12), 3547–3562.
Pavone, P., et al. (2022). Long-term outcomes in West syndrome. Brain Sciences, 12(8), 1050.
Raga, S., Specchio, N., Rheims, S., et al. (2023). Pathophysiology of epileptic spasms. Epilepsia, 64(S1), S10–S21.
Riikonen, R. (2022). Long-term prognosis of infantile spasms. Epileptic Disorders, 24(1), 1–13.
Shellhaas, R. A., Wusthoff, C. J., Tsuchida, T. N., et al. (2023). Electroencephalography in infantile spasms. Journal of Clinical Neurophysiology, 40(3), 190–200.
Smith, M. S., Matthews, R., Rajnik, M., & Mukherji, P. (2024). Infantile epileptic spasms syndrome (West syndrome). StatPearls Publishing.
Specchio, N., & Curatolo, P. (2023). Developmental and epileptic encephalopathies: Clinical features. The Lancet Neurology, 22(4), 335–348.
Stafstrom, C. E., & Carmant, L. (2022). Seizures and epilepsy: An overview. The Lancet, 399(10343), 2310–2320.
Symonds, J. D., Zuberi, S. M., & Stewart, K. (2022). Advances in epilepsy gene discovery and implications for West syndrome. Nature Reviews Neurology, 18(4), 210–222.
Widjaja, E., Li, B., Schinkel, C., et al. (2022). Prognostic factors in infantile spasms. Epilepsia, 63(3), 612–624.
Widjaja, E., Go, C., & McCoy, B. (2023). Ketogenic diet in infantile spasms. Epilepsy Research, 195, 107200.
Wilmshurst, J. M., et al. (2022). Diagnostic approach to epilepsy syndromes. Epileptic Disorders, 24(2), 140–155.
Wilmshurst, J. M., Gaillard, W. D., Vinayan, K. P., et al. (2022). Summary of recommendations for the management of infantile seizures. Epilepsia, 63(8), 1880–1895.
Wirrell, E. C., Shellhaas, R. A., Joshi, C., et al. (2022). How should children with West syndrome be evaluated? Epileptic Disorders, 24(1), 14–33.